Синдром Уивера (OMIM #277590) — это генетическое заболевание, которое обуславливает быстрый избыточный рост на всем протяжении периода роста, сочетающийся с аномалиями развития скелета и внутренних органов и в 85% случаев с умственной отсталостью.
В 1974 г. американские генетики, возглавляемые известным ученым, профессором медицинской и молекулярной генетики Медицинской школы Университета Индианы D. Weaver, впервые опубликовали случай данной патологии у двух мальчиков [1]. До настоящего времени в мире описано не более 50 наблюдений этого синдрома. Из ныне живущих больных с синдромом Уивера известна 22-летняя турчанка Румейса Гелги (Rumeysa Gelgi), которая занесена в книгу рекордов Гиннеса как самая высокая женщина в мире. Ее рост составляет 2 метра 15 см. В РНПЦ «Мать и дитя» Минздрава Республики Беларусь в 2013 г. опубликовано 5 случаев наблюдения данного синдрома [2]. В отечественной литературе найдено описание клинического примера 5-летней девочки, наблюдавшейся в Московском НИИ педиатрии и детской хирургии Минздрава РФ в 2000 г. [3].
Тип наследования синдрома Уивера до сих пор остается неясным. Большинство описанных случаев были спорадическими. Проявления синдрома Уивера характеризуются вариабельной экспрессивностью и большей частотой среди лиц мужского пола.
Диагностика заболевания состоит из следующего симптомокомплекса: высокие показатели физического развития (SDS роста > 2), типичные краниофасциальные симптомы, низкий хриплый голос, изменения опорно-двигательного аппарата, поражение органов зрения, возможна задержка психомоторного и речевого развития, часто нормальный уровень соматомедина С и гормона роста, костный возраст соответствует паспортному или опережает его.
Генетическая основа синдрома Уивера длительное время оставалась неизвестной. В 2003 г. J. Douglas с соавт. описали мутации в гене NSD1, отвечающем за развитие синдрома Сотоса, у 3 пациентов из 7 с диагнозом «синдром Уивера» [4]. Однако в 2012 г. было проведено полное секвенирование экзома неродственным пациентам с синдромом Уивера и в результате выявлены мутации в гене EZH2, расположенном на 7-й хромосоме [5].
Дифференциальная диагностика проводится с другими заболеваниями, сопровождающимися макросомией и аномалиями развития: в первую очередь с синдромом Сотоса, синдромом Видемана–Беквита, синдромом МОМО, синдромом Симпсона–Голаби–Бемеля, полисомией Y, синдромом Вермера, синдромом Карнея, синдромом Клайнфельтера, синдромом Марфана, гомоцистинурией, синдромом Пайла, синдромом Маршалла–Смита и другими.
Клиническое наблюдение
Мальчик поступил в педиатрическую клинику МОНИКИ впервые в возрасте 15 лет с жалобами на высокий рост (за последний год прирост 15 см), увеличение размера ноги за год с 44 до 48, деформацию позвоночника и грудной клетки, ухудшение зрения.
Ребенок от второй беременности (первый ребенок здоров), протекавшей на фоне повторных угроз прерывания на ранних сроках. Роды срочные, самопроизвольные. Масса при рождении 4200 г, длина 57 см. С рождения физическое развитие гармоничное, высокое. Психомоторное развитие и половое развитие соответствуют возрасту. Наблюдается офтальмологом по поводу гиперметропии высокой степени обоих глаз, хирургом-ортопедом по поводу нарушения осанки, деформации грудной клетки, плоскостопия, челюстно-лицевая хирургия по поводу микрогнатии (получает лечение в виде дистракционного остеосинтеза нижней челюсти с двух сторон) и ортодонтом по поводу нарушения прикуса и роста зубов. Рост отца и матери ребенка 172 см. У родственников мужского пола по линии матери рост 180 см — 2 м.
С рождения отмечались большие прибавки роста. При обследовании в 2 года — физическое развитие на 4 года. Общеклинические анализы в норме. Гормон роста в пределах референсных значений. Костный возраст на 3–4 года. МРТ головного мозга — без патологии. Ребенок и в детском саду, и в школе был значительно выше своих сверстников.
С 2012 г. появились жалобы на головокружение, головную боль, периодический акроцианоз, потерю сознания при изменении положения тела. При мониторинге артериального давления выявлена склонность к гипотонии. Предъявляемые ребенком жалобы объяснялись отставанием развития сердечной мышцы и сосудистой системы по сравнению со скоростью роста костей, тогда как сама причина такого стремительного роста оставалась неясной.
В 2017 г. мальчик поступил в педиатрическое отделение МОНИКИ с направительным диагнозом: «Синдром высокорослости».
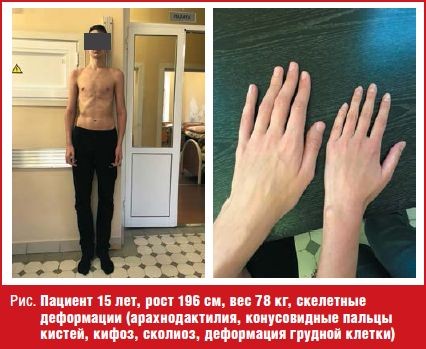
При поступлении рост мальчика рост 196 см, вес 78 кг. Обращали на себя внимание: грубый хриплый голос, лицевые дизморфии (антимонголоидный разрез глаз, удлиненные глазные щели, широкий и короткий нос, широкий длинный фильтр, ретромикрогнатия, большие диспластичные ушные раковины), скелетные деформации (арахнодактилия, конусовидные пальцы кистей, кифоз, сколиоз, деформация грудной клетки) (рис.). По лабораторным данным: общий анализ крови, мочи, кала, развернутый биохимический анализ крови, гормональный профиль (гормоны щитовидной железы, гипофиза, надпочечников, половые гормоны), исследование фосфорно-кальциевого обмена — показатели в пределах референсных значений. ЭКГ, Эхо-КГ, УЗИ брюшной полости, забрюшинного пространства и щитовидной железы — без особенностей. Уровень артериального давления в пределах возрастной нормы. МРТ головного мозга и гипоталамо-гипофизарной области прицельно — без патологии. Рентгенограмма кистей — костный возраст соответствует 15,5–16 годам. Осмотрен совместно с узкими специалистами (эндокринолог, генетик): SDS роста +3,4. Половое развитие соответствует возрасту. Высокорослость. Марфаноподобный синдром.
В результате комплексного обследования в клинике эндокринная причина избыточного роста ребенка была исключена, и тогда было принято решение провести углубленное генетическое обследование. Проведен поиск мутаций, ассоциированных с синдромом Марфана и другими наследственными заболеваниями со сходными фенотипическими проявлениями. В результате молекулярно-генетического анализа у мальчика выявлена мутация в 17-м экзоне гена EZH2 в гетерозиготном состоянии. Мутация в гене EZH2 является типичной для синдрома Уивера, однако выявленная у пациента мутация ранее не была описана.
Таким образом, учитывая фенотипические особенности пациента и наличие мутации в гене EZH2, был установлен диагноз «синдром Уивера».
Спустя год ребенок обследован в клинике повторно. Рост 2,01 м (годовая прибавка роста 5 см), вес 86 кг. Размер обуви остался без изменений. В ходе обследования общеклинические анализы, данные гормонального профиля, форфорно-кальциевого обмена, УЗИ брюшной полости, Эхо-КГ, мониторинг артериального давления, МРТ головного мозга, денситометрия в режиме позвоночник и все тело — без патологии. Костный возраст соответствует 16–16,5 годам.
Заключение
Несмотря на то, что все генетически детерминированные синдромы высокорослости встречаются крайне редко, характеризуются схожими проявлениями, не имеют специфического лечения, пациентам необходимо проводить комплексное генетическое обследование. В зависимости от генетического дефекта, различается спектр клинических проявлений различных синдромов, что позволяет прогнозировать в дальнейшем течение заболевания и осуществлять профилактические мероприятия по развитию некоторых осложнений. Так, например, учитывая, что мутации в гене EZH2 являются причиной синдрома Уивера, а ген EZH2 является белком гомеобокс группы, который участвует в поддержании гомеостаза, деления и дифференцировки клеток, соответственно, патологическая экспрессия этого гена у больных синдромом Уивера может быть причиной нейродегенеративных заболеваний и некоторых форм рака, например, нейробластомы. Кроме того, в гене EZH2 описаны мутации в соматических клетках при лейкемии, В-клеточных лимфомах и других онкологических заболеваниях крови, что обуславливает некоторую предрасположенность пациентов с синдромом Уивера к онкологическим заболеваниям [5–7]. Эти сведения могут помочь лечащему врачу в определении тактики наблюдения пациента и осуществления профилактических мероприятий.
Литература
- Weaver D. D., Graham C. B., Thomas I. T., Smith D. W. A new overgrowth syndrome with accelerated skeletal maturation, unusual facies, and camptodactyly // J. Pediatr. 1974; 84: 547–552.
- Ильина Е. Г., Ершова-Павлова А. А. Синдром Вивера в Беларуси // Здравоохранение (Минск), 2013; 3: 62–65.
- Казанцева Л. З., Семячкина А. Н., Новиков П. В., Новикова И. М., Добрынина Э. В. Синдром Вивера у детей // Российский вестник перинатологии и педиатрии. 2000; 2: 55–57.
- Douglas J. et al. NSD1 mutations are the major cause of Sotos syndrome and occur in some cases of Weaver syndrome but are rare in other overgrowth phenotypes // Am J Hum Genet. 2003, Jan; 72 (1): 132–143.
- Gibson, W. T., Hood R. L. et al. Mutations in EZH2 cause Weaver syndrome // Am. J. Hum. Genet. 2012, 90: 110–118.
- Качанов Д. Ю., Шаманская Т. В., Шевцов Д. В. и др. Генетическая предрасположенность к нейробластоме у детей: собственные данные и обзор литературы // Онкопедиатрия. 2016; 3 (4): 277–287.
- Chase A., Cross N. C. Aberrations of EZH2 in cancer // Clin Cancer Res. 2011, May 1; 17 (9): 2613–2618.
Д. А. Карташова
Ю. Ю. Коталевская, кандидат медицинских наук
ГБУЗ МО МОНИКИ им. М. Ф. Владимирского, Москва
1 Контактная информация: bta2304@mail.ru
Синдром Уивера у ребенка 16 лет (описание клинического случая)/ Т. А. Бокова, Д. А. Карташова, Ю. Ю. Коталевская
Для цитирования: Лечащий врач № 1/2019; Номера страниц в выпуске: 12-13
Теги: синдром Уивера, физическое развитие, отклонения
Купить номер с этой статьей в pdf