Резюме
Введение. Согласно российским исследованиям, средний возраст постановки диагноза «прогрессирующая мышечная дистрофия Дюшенна» составляет 7-8 лет. Это связано с тем, что, с одной стороны, миодистрофия Дюшенна – орфанная патология и врач за всю свою клиническую практику может ни разу не увидеть ее. С другой – клиническая картина заболевания многолика и не имеет ярко выраженной симптоматики на начальных этапах развития. Часто врач ошибается с диагнозом, принимая следствие заболевания за его источник или рассматривая лишь одно из проявлений болезни и при этом не видя всего симптомокомплекса и ее первопричину. Так, одним из проявлений заболевания является повышение печеночных ферментов – трансаминаз (аланинаминотрансферазы, аспартатаминотрансферазы) и лактатдегидрогеназы. Около 50% всех ошибочных диагнозов при миодистрофии Дюшенна связаны с повреждением печени. Такие дети могут годами наблюдаться у инфекционистов или детских гастроэнтерологов с неуточненными диагнозами, проходя все новые и новые, подчас инвазивные, исследования.
Заключение. В настоящее время многим пациентам с миодистрофией Дюшенна доступна патогенетическая терапия. К сожалению, она не возвращает утраченные функции, но может сохранить имеющиеся и клинически перевести агрессивную форму миодистрофии Дюшенна в более мягко протекающую форму Беккера. Именно поэтому так важно информировать врачей как первичного педиатрического звена, так и детских гастроэнтерологов и инфекционистов о механизмах внепеченочного повышения трансаминаз и алгоритмах диагностики миодистрофий.
Конфликт интересов. Автор статьи подтвердила отсутствие конфликта интересов, о котором необходимо сообщить.
Мышечная дистрофия Дюшенна (МДД) — наследственное, рецессивное, неуклонно прогрессирующее нервно-мышечное заболевание, сцепленное с Х-хромосомой и вызванное патогенными вариантами в гене DMD. Частота встречаемости МДД составляет около 1 на 5000 новорожденных мальчиков. Причиной развития заболевания являются различные патогенные варианты в гене DMD, которые приводят к недостатку или полному отсутствию белка дистрофина. Среди них, согласно российским данным, выделяют крупные делеции (≈51% от общего количества мутаций), точковые замены (≈35%) и дупликации (≈14%) [1]. В большинстве случаев патогенный вариант передается от матери ребенку, однако известно, что в трети случаев он может возникать и спонтанно (de novo) [2].
Продуктом гена DMD является белок дистрофин, который экспрессируется преимущественно в мышечной ткани (скелетные мышцы, сердце, кишечник и диафрагма), но также известно, что некоторые его изоформы присутствуют в центральной нервной системе и сетчатке [3]. Количество дистрофина в поперечнополосатых мышцах составляет около 0,002%, однако, несмотря на такую незначительную долю от массы белка всей мышцы, его отсутствие является фатальным для ребенка. Этот белок связывает внутренний цитоскелет с сарко- и дистрогликанами в мембране и внеклеточном матриксе, обеспечивает механическую и структурную стабильность мембраны мышечных волокон при их сокращении. Дистрофин также является амортизатором, обеспечивающим возвращение мышцы в исходное состояние после сокращения. При прогрессировании заболевания мышечные волокна разрушаются и замещаются фиброзной и жировой тканью [2]. МДД поражает преимущественно мальчиков и приводит к нарушению функций органов-мишеней, что в свою очередь служит причиной ранней инвалидизации и смерти на третьем десятилетии при отсутствии вовремя начатого лечения [2].
Одной из особенностей данного заболевания является раннее проявление клинических симптомов, которые могут быть приняты за нарушение психомоторного и речевого развития. Симптомы МДД можно разделить на две большие группы.
1. Моторные нарушения:
- неуклюжесть походки и частые падения;
- мышечная гипотония;
- боли в мышцах;
- снижение выносливости;
- невозможность прыгать;
- изменение походки;
- трудности при ходьбе по лестнице;
- применение приемов Говерса (вставание лесенкой по себе или окружающим предметам) при подъеме с пола или с корточек;
- потеря двигательных навыков.
2. Немоторные нарушения:
- поведенческие нарушения;
- задержка речи;
- нарушения артикуляции;
- задержка формирования когнитивных функций;
- нарушения обучения и удержания внимания;
- кардиомиопатия;
- изменения в биохимическом анализе крови – повышение трансаминаз: аланинаминотрансферазы (АЛТ), аспартат-аминотрансферазы (АСТ), креатинфосфокиназы (КФК), лактатдегидрогеназы (ЛДГ) [4].
К сожалению, довольно часто в реальной клинической практике основной жалобой законных представителей несовершеннолетнего является нарушение походки – от поздних сроков начала ходьбы до сложностей с хождением по лестнице. В этом случае родители в первую очередь обращаются к ортопеду-травматологу, который видит вальгусную деформацию и старается скорректировать ее, зачастую не понимая, что это лишь следствие основного заболевания.
Другим примером является задержка речевого или психоречевого развития. С этими жалобами родители чаще всего обращаются к неврологу или логопеду, который не всегда оценивает состояние пациента полностью и назначает лишь симптоматическую терапию.
Еще одним примером является мышечная гипотония. При такой жалобе участковый педиатр с большей вероятностью направит ребенка на массаж. Однако и в этом случае результаты окажутся хуже ожидаемых, так как массаж с использованием стимулирующих техник при МДД строго противопоказан и приведет лишь к прогрессированию заболевания.
Но чаще всего во время диспансеризации или госпитализации ребенка по любому поводу проводят стандартное биохимическое исследование крови, которое включает в себя исследование печеночных трансаминаз (АЛТ и АСТ). Традиционно повышение АЛТ и АСТ врачи связывают с поражением печени, так как это маркеры гепатоцеллюлярного повреждения, в связи с чем ребенка направляют на консультацию к детскому инфекционисту или гастроэнтерологу. И начинаются долгие, изнурительные для всех участников поиски поражения печени. Учитывая российскую действительность, очередь к узким специалистам подобного плана может растянуться на недели и месяцы. В дальнейшем – продолжительное исследование и поиск причин повышения уровня печеночных ферментов, который растягивается на годы. Согласно последним исследованиям около половины ошибочных диагнозов приходится на гепатиты неясного генеза [5]. Вместе с тем повышение печеночных трансаминаз при МДД имеет внепеченочное происхождение [6, 7].
Показано, что различные нейромышечные заболевания, такие как мышечные дистрофии, воспалительные и метаболические миопатии, могут привести к повышению уровня АСТ, АЛТ, а также ЛДГ и КФК [8]. При МДД происходит нарушение целостности мембраны миоцитов из-за недостаточности или полного отсутствия белка дистрофина, и содержимое клетки изливается в межклеточное пространство. Оттуда трансаминазы и попадают в кровеносное русло [8, 9].
На ранних стадиях развития МДД повышение АЛТ и АСТ может быть единственным проявлением заболевания, когда еще нет основных клинических симптомов болезни или когда присутствуют только едва заметные признаки в виде плохой переносимости физической нагрузки и неуклюжести [10, 11]. Хотя многие другие состояния (такие как вирусный гепатит, аутоиммунные нарушения, неалкогольный стеатогенный гепатит и целиакия, болезнь Вильсона и др.) могут являться причиной повышения печеночных ферментов, мышечные заболевания также следует исключать на ранней стадии дифференциальной диагностики у ребенка с изолированным повышением АСТ и АЛТ. Таким образом, случайное обнаружение повышенного уровня АЛТ/АСТ без других признаков поражения печени может быть характерным признаком заболевания мышц у многих детей и дает возможность ранней диагностики грозного нейромышечного заболевания.
Существует простой и эффективный алгоритм дифференциальной диагностики гипертрансаминаземии гепатоцеллюлярного и нейромышечного генеза (рис.).
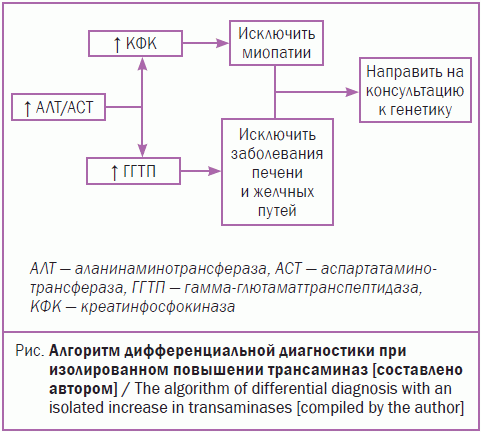
Представленный алгоритм дифференциальной диагностики важен для педиатров первичного звена. Проведя несложное дообследование, врач точно будет понимать, к какому специалисту необходимо отправить ребенка – к неврологу или гастроэнтерологу/инфекционисту. Внедрение подобного алгоритма позволит значительно сократить время постановки диагноза и рекомендации терапии, что, в свою очередь, поможет сохранить не только полноценного члена общества, но и снизить социальную нагрузку и сохранить качество жизни семьи.
Об этой схеме стоит также помнить и детским гастроэнтерологам, и специалистам по инфекционным болезням, так как часто именно к ним попадают дети с повышенным уровнем трансаминаз.
Очевидно, что на участковых педиатрах лежит большая ответственность – наблюдая за развитием каждого ребенка, они первыми могут увидеть отклонения от принятых норм, обратить внимание на тревожные жалобы мам. Ведь чтобы снять подозрения или успокоить родителей, нет необходимости в большом и дорогостоящем обследовании. Достаточно взять несколько биохимических показателей и получить дальнейший вектор диагностического поиска при выходе полученных результатов за пределы референтных значений.
Облигатным маркером МДД является КФК. При МДД уровни КФК в крови могут повышаться в 10-100 раз. Именно это исследование позволяет выявить заболевание на досимптомной стадии, когда лечение даст наилучшие результаты. При выявлении значительного повышения (более 1000 Ед/л) КФК у мальчика следует заподозрить МДД и продолжить обследование для уточнения диагноза.
Согласно клиническим рекомендациям, диагноз МДД устанавливается на основании проведения молекулярно-генетического исследования. Золотым стандартом здесь является мультиплексная амплификация лигированных зондов (MLPA). Эта реакция направлена на выявление крупных делеций и дупликаций гена DMD, которые в сумме составляют ≈75% всех встречающихся мутаций. Выявить малые и точечные мутации поможет массовое параллельное секвенирование экзома (МПС). МПС позволяет обнаружить 98% патогенных вариантов в гене. При их выявлении таким способом необходимо подтверждение методом прямого секвенирования по Сенгеру гена DMD [2].
Хотя диагностика генетических заболеваний – дорогостоящее исследование, для пациентов с подозрением на МДД оно проводится бесплатно. ФГБНУ «Медико-генетический научный центр имени академика Н. П. Бочкова» проводит как диагностику этого заболевания в лаборатории ДНК-диагностики, так и медико-генетическое консультирование в консультативном отделении на бюджетной основе. Для проведения исследования необходимо соблюсти следующие условия:
- пациент мужского пола;
- житель России или СНГ;
- уровень КФК в анализе крови более 1000 Ед/л.
На сайте МГНЦ можно найти всю информацию об этой и многих других диагностических программах. Лечащему врачу необходимо связаться с МГНЦ им. Н. П. Бочкова по телефону, указанному на сайте программы диагностики, и следовать дальнейшему алгоритму действий, который будет сообщен оператором. Чем раньше это будет сделано, тем быстрее пациент получит необходимое лечение [12].
Почему же так важно начать лечение на бессимптомной стадии? Как уже упоминалось ранее, при МДД происходит жировое перерождение и фиброзное замещение мышц из-за отсутствия белка дистрофина в них. При этом с каждым днем количество патологически измененных мышц увеличивается, снижая продолжительность жизни пациента. Помимо скелетных мышц, страдает сердечно-сосудистая система, в большей степени сердце, дыхательная система (происходит снижение жизненной емкости легких), изменениям подвергается и опорно-двигательный аппарат – контрактуры в суставах прогрессируют по мере развития заболевания. Все вышеперечисленные изменения напрямую влияют на качество жизни не только самого пациента, но и его семьи. Без должного лечения ребенок не сможет стать самостоятельным и активным членом общества, а его законные представители будут вынуждены жертвовать карьерой и репродуктивными планами во имя жизни больного с МДД. Предлагаемая этиопатогенетическая терапия на современном этапе пока не способна навсегда вылечить пациента, однако она может значительно увеличить продолжительность его жизни и дать шанс, что она будет полноценной.
Одним из вариантов патогенетической терапии является аталурен. Этот препарат может помочь только тем пациентам, у которых МДД была вызвана нонсенс-мутацией в гене DMD. Механизм его действия направлен на продолжение синтеза белка дистрофина, несмотря на наличие стоп-кодона, который обычно прерывает процесс трансляции.
Другим вариантом воздействия на болезнь является экзон-скиппинг-терапия. Метод экзон-скиппинга (ЭС) заключается в обеспечении возможности обойти делетированный экзон или экзоны в процессе сплайсинга и восстановить рамку считывания. Таким образом, из зрелой мРНК и, как следствие, из белка данная мутация будет исключена. Механизм ЭС заключается в связывании антисмысловых олигонуклеотидов, малых синтетических молекул РНК, с определенными участками целевой пре-мРНК, что и позволяет исключить из процесса сплайсинга определенные участки РНК [13].
В настоящее время российским пациентам доступны препараты, которые направлены на пропуск следующих экзонов: 53, 45, 51. Несмотря на высокую стоимость препаратов для патогенетической терапии МДД, важно донести до родителей, что в настоящее время есть возможность получать эти препараты за счет средств фондов, которые закупают их для пациентов. Кроме того, из-за того что не все препараты зарегистрированы на территории РФ, их практически невозможно получить по ОМС, а фонд «Круг добра» дает такую возможность.
Исходя из всего вышесказанного, можно сделать вывод о том, что жизнь пациентов с МДД поддается изменениям исключительно в лучшую сторону. Основой продления качественной и долгой жизни является ранняя диагностика, своевременное назначение симптоматической терапии и регулярное выполнение комплекса упражнений для детей с МДД, которое полностью ложится на плечи родителей маленького пациента. Лишь при соблюдении этих трех условий своевременное назначение патогенетической терапии может значимо повысить качество жизни пациентов и их семей, продлить годы амбулаторного периода болезни, предоставить обществу полноценного члена. Врачам важно и нужно уметь замечать не только симптомы этого заболевания, но и уметь заподозрить его еще на бессимптомной стадии, опираясь на свой опыт, знания, интуицию и результаты биохимического исследования крови. Поэтому в заключение статьи хотим еще раз напомнить участковым педиатрам, которые стоят на передовой, охраняя здоровье и благополучие наших маленьких граждан: если у вас появился пациент со случайно выявленным повышением трансаминаз или ЛДГ, проверьте у него уровень активности КФК и гамма-глютаматтранспептидазы! Это поможет составить правильный маршрут дальнейшего поиска причин таких изменений. Если КФК повышен до 1000 Ед/л и более, можете сами назначить тестирование гена
Литература/References
- Зинина Е. В., Булах М. В., Рыжкова О. П., Щагина О. А., Поляков А. В. Изменение спектра выявленных мутаций в гене DMD в зависимости от методических возможностей лаборатории. Нервно-мышечные болезни. 2023; 13 (1): 33-43. / Zinina E. V., Bulakh M. V., Ryzhkova O. P., Shchagina O. A., Polyakov A. V. Change in the spectrum of detected mutations in the DMD gene depending on the methodological capabilities of the laboratory. Nervno-myshechnye bolezni. 2023; 13 (1): 33-43. (In Russ.)
- Клинические рекомендации. Прогрессирующая мышечная дистрофия Дюшенна. Прогрессирующая мышечная дистрофия Беккера. МЗ. 2023. / Clinical recommendations. Progressive Duchenne muscular dystrophy. Becker's progressive muscular dystrophy. MoH. 2023. (In Russ.)
- Shin’ichi Takedaa, Clemensb P. R., Hoffman E. P. Exon-Skipping in Duchenne Muscular Dystrophy. Journal of Neuromuscular Diseases. 2021; 8: S343-S358.
- Ciafaloni E., Fox D. J., Pandya S., et al. Delayed diagnosis in Duchenne muscular dystrophy: data from the Muscular Dystrophy Surveillance, Tracking, and Research Network (MD STARnet). J Pediatr. 2009; 155: 380-385.
- Гремякова Т. А., Гремякова О. И. «Невидимые» мальчики. Редкие болезни в России. 2021; 19. / Gremyakova T. A., Gremyakova O. I. The «invisible» boys. Redkie bolezni v Rossii. 2021; 19. (In Russ.)
- Korones D. N., Brown M. R., Palis J. Liver Function Tests Are Not Always Tests of Liver Function. American. Journal of Hematology. 2001; 66: 46-48.
- Wright M. A., Yang M. L., et al. Consider Muscle Disease in Children with Elevated Transaminase. JABFM. 2012; 4 (25).
- McMillan H. J., Gregas M., Darras B. T., Kang P. B. Serum transaminase levels in boys with Duchenne and Becker muscular dystrophy. Pediatrics. 2011; 127: e132-136.
- Vajro P., Maddaluno S., Veropalumbo C. Persistent hypertransaminasemia in asymptomatic children: a stepwise approach. World J Gastroenterol. 2013; 19: 2740-2751.
- Pearson C. M., Chowdhury S. R., Fowler W. M. Jr., et al. Studies of enzymes in serum in muscular dystrophy. II. Diagnostic and prognostic significance in relatives of dystrophic persons. Pediatrics. 1961; 28: 962-970.
- Thomson W. H. Serum enzyme studies in acquired disease of skeletal muscle. Clin Chim Acta. 1971; 35: 193-199.
- Электронный ресурс. Научные диагностические программы (med-gen.ru) https://med-gen.ru/spetcialistam/nauchnye-diagnosticheskie-programmy/. Дата обращения 21.02.2024. Website. Scientific diagnostic programs (med-gen.ru) https://med-gen.ru/spetcialistam/nauchnye-diagnosticheskie-programmy/. (In Russ.) Acessed: 21.02.2024.
- Яковлев И. А., Деев Р. В., Соловьева В. В. и др. Пред- и посттранскрипционная модификация генетической информации в программе лечения мышечных дистрофий. Гены & клетки. 2016; 2 (XI). Yakovlev I. A., Deev R. V., Solovyеva V. V., et al. Pre- and posttranscriptional genetic information modification in muscular dystrophy treatment. Geny & kletki. 2016; 2 (XI). (In Russ.)
Федеральное государственное бюджетное научное учреждение Медико-генетический научный центр имени академика Н. П. Бочкова Министерства науки и высшего образования Российской Федерации; 115522, Россия, Москва, ул. Москворечье, 1
Сведения об авторе:
Шаркова Инна Валентиновна, д.м.н., невролог, ведущий научный сотрудник научно-консультативного отдела ФГБНУ «Медико-генетический центр имени академика Н. П. Бочкова» Министерства науки и высшего образования Российской Федерации, 115522, Россия, Москва, ул. Москворечье, 1; sharkova.inna@gmail.com
Information about the author:
Inna V. Sharkova, Dr. of Sci. (Med.), Neurologist, Leading researcher of the scientific advisory at the Federal State Budgetary Scientific Institution Research Center named after Academician N. P. Bochkov of the Ministry of Science and Higher Education of the Russian Federation; 1 Moskvorechye str., Moscow, 115522, Russia; sharkova.inna@gmail.com
Повышение трансаминаз. Что, если не печень?/ И. В. Шаркова
Для цитирования: Шаркова И. В. Повышение трансаминаз. Что, если не печень? Лечащий Врач. 2024; 3 (27): 67-70. https://doi.org/10.51793/OS.2024.27.3.011
Теги: прогрессирующая мышечная дистрофия, диагностика, алгоритмы
Купить номер с этой статьей в pdf