Оригинальная статья: Blázquez E. et al. Insulin in the brain: its pathophysiological implications for States related with central insulin resistance, type 2 diabetes and Alzheimer's disease. Front Endocrinol (Lausanne). 2014;5:161. DOI:10.3389/fendo.2014.00161. PMID: 25346723
Авторы: Enrique Blázquez, Esther Velázquez, Verónica Hurtado-Carneiro and Juan Miguel Ruiz-Albusac
Перевод: Ирина Буянова. Редакция: Илья Левашов.
Аннотация
Инсулин может попадать в мозг как из периферических тканей, так и синтезироваться клетками центральной нервной системы (ЦНС). Молекулярные механизмы действия инсулина в ЦНС не отличаются от периферического действия гормона. Результаты последних исследований демонстрируют нейротрофический, нейромодуляторный и нейропротективный эффекты инсулина и его основополагающую роль в регуляции гомеостаза глюкозы, репродуктивной и когнитивных функций, пищевого поведения, а также пролиферации и дифференцировке клеток.
Нарушение механизмов инсулиновой сигнализации в мозге лежит в основе патогенеза центральной инсулинорезистентности, сахарного диабета 2 типа (СД2) и болезни Альцгеймера (БА). Кроме того, появляется все больше данных о связи между нарушением метаболизма глюкозы в ЦНС и риском БА.
Общими звеньями патогенеза БА и СД2 являются митохондриальная дисфункция и нарушение энергетического гомеостаза, оксидативный стресс, образование модифицированных липопротеинов низкой плотности (ЛПНП), снижение активности O-GlcNAc трансферазы, формирование амилоидных бляшек, изменение метаболизма бета-амилоидного пептида (Aβ) и повышение фосфорилирования тау-белка.
Введение
Поддержание гомеостаза глюкозы в периферических тканях – одна из основных функций инсулина. В последнее время появляется все больше данных о наличии рецепторов к инсулину (ИР) в разных отделах ЦНС и их роли в регуляции важнейших физиологических функций, включая нейрональное развитие, метаболизм глюкозы, пищевое поведение и когнитивные процессы, а также в патогенезе нейродегенеративных заболеваний (1).
Как инсулин попадает в мозг
Присутствие инсулина в нервной ткани впервые было описано в работе Havrankova et al. в 1978 году и подтверждено многочисленными исследованиями на животных (2, 3). Исследователи показали, что уровень инсулина в ЦНС не зависит от концентрации инсулина в периферических тканях и превышает его в 10-100 раз (4).
Присутствие инсулина в цереброспинальной жидкости (ЦСЖ) служит биомаркером проницаемости гематоэнцефалического барьера (ГЭБ), который определяет эффективность проникновения гормона в ЦНС и препятствует его быстрому выведению (5, 6). Считается, что основным источником инсулина в ЦНС служат специфические популяции нейронов, хотя, как отмечает Havrankova et al. (2), не стоит исключать проникновение инсулина из периферического кровотока и через ГЭБ.
Инсулин периферического происхождения
В 1967 году Margolis и Altszuler впервые высказали гипотезу о наличии механизма, опосредующего транспорт инсулина через ГЭБ (7). В эксперименте на крысах авторы работы показали, что внутривенное введение инсулина вызывает незначительное повышение его уровня в ЦСЖ. Это привело к предположению, что инсулин может проникать через ГЭБ посредством рецепторзависимого транспорта (8). Выраженное повышение уровня инсулина в крови и незначительное увеличение его концентрации в ЦСЖ в ответ на системное введение гормона указывает на наличие специальной транспортной системы, позволяющей доставлять инсулин через ГЭБ. Хотя до сих пор неизвестно, являются ли переносчики инсулина и ИР в ЦНС представителями одного семейства белков, данные многочисленных исследований демонстрируют (9) сходные физико-химические свойства (насыщаемость, специфичность, афинность, кинетику распада и иммунонейтрализацию) этих белков (10, 11). С другой стороны, активность транспортеров ГЭБ может меняться в зависимости от региональных различий в проницаемости к инсулину и уровню гормона в крови. Наиболее высокое содержание инсулина обнаружено в мосте, продолговатом мозге и гипоталамусе; самый низкий уровень гормона показан в нейронах затылочной коры и тал��муса (12). Транспорт инсулина через ГЭБ регулируется различными физиологическими факторами, например, действием глюкокортикоидных гормонов (13), и обладает высокой чувствительностью к изменению энергетического гомеостаза при голодании (14), ожирении (15), гибернации (16), а также в процессе старения и у пациентов с СД и БА (17, 18).
Инсулин центрального происхождения
Биосинтез инсулина в ЦНС хорошо изучен и был продемонстрирован в ряде экспериментальных исследований.
Определение уровня С-пептида в мозге
В исследованиях post mortem показано высокое содержание иммунореактивного инсулина и С-пептида в различных структурах мозга, в несколько раз превышающее их содержание в периферической крови (19). Уровень С-пептида в сыворотке крови и структурах мозга значительно снижается после 72 часов голодания и повышается в ответ на пероральное введение глюкозы (20). Прямое доказательство связи между уровнем С-пептида и снижением количества ИР в ЦНС было получено в исследовании с участием пожилых пациентов с БА, у которых наиболее высокий уровень С-пептида был обнаружен в коре головного мозга (21).
С помощью метода гибридизации in situ удалось выделить мРНК инсулина из нейронов перивентрикулярного ядра гипоталамуса крыс (22). Кроме того, мРНК инсулина II присутствует в мозге крыс на всех этапах развития, что указывает на раннюю экспрессию гена-предшественника в мозге (23). У новорожденных кроликов показано наличие мРНК инсулина в CA1 и CA3 областях гиппокампа, зубчатой извилине и клетках гранулярного слоя обонятельной луковицы (24).
Экспериментальные подходы к работе с культурами клеток мозга
Многочисленные данные о механизмах синтеза инулина в ЦНС были получены в исследованиях на культурах клеток. Показано, что инкубация нейронов в растворе циклогексимида приводила к 80%-му снижению количества инсулин-иммунореактивных клеток в мозге крыс (25). В эмбриональных нейронах мышей было показано наличие двух форм иммунореактивного инсулина (ИРИ). Под действием трипсина про-инсулин расщепляется на компоненты, схожие по структуре с панкреатическим инсулином (26). Данные иммуногистохимического анализа и гибридизации in situ демонстрируют способность эмбриональных нейронов экспрессировать и выделять инсулин-подобные мРНК и инсулин-подобные белки, схожие по структуре с инсулином периферического происхождения (27). В мозге млекопитающих синтез иммунореактивного инсулина происходит в только нейронах ЦНС, но не в астроцитах (24).
Молекулярные механизмы, участвующие в регуляции образования и секреции инсулина в ЦНС, сходны с механизмами синтеза инсулина в периферических тканях. Одним из общих звеньев центрального и периферического путей образования инсулина является деполяризация, вызванная активацией АТФ-чувствительных K+ каналов (28). Связывание гормонов или глюкозы с рецепторами на поверхности клеточных мембран вызывает деполяризацию и стимулирует высвобождение инсулина. В культурах нейронов (но не астроцитов) циклогексимид ингибирует экзоцитоз, индуцируемый деполяризацией (29). Кроме того, в мозге взрослых крыс повышение мембранного потенциала способствует высвобождению инсулина.
Эффективность экзоцитоза инсулина во многом зависит от внутриклеточного содержания уровня кальция. Кальций-зависимый экзоцитоз специфичен для синаптических везикул нервных окончаний, что подтверждает данные о синтезе инсулина в нейронах ЦНС и его хранении в составе синаптических везикул (30).
Секреция инсулина из синаптосом повышается в ответ на увеличение уровня глюкозы, а добавление ингибитора гликолитического метаболизма – йодацетиловой кислоты (ЙК) – в культуры клеток мозга вызывает снижение высвобождения ИРИ на 50%, что подчеркивает регуляторную роль метаболизма глюкозы в секреции инсулина в ЦНС (31).
Таким образом, результаты последних исследования подтверждают наличие центрального пути синтеза инсулина (32).
Влияние инсулина на эндотелиальные клетки и ГЭБ
ГЭБ образован особым типом эндотелиальных клеток (33), которые формируют физический барьер между клетками мозга и кровотоком (17). Поверхность клеток эндотелия содержит инсулин-связывающие сайты, которые выполняют транспортную и рецепторную функции (34). Эти инсулин-связывающие сайты играют ключевую роль в регуляции внутриклеточных каскадов в клетках ГЭБ и инсулиновой сигнализации, а также способствуют повышению эффективности транспорта тирозина и триптофана (35), азидотимидина (36) и лептина (37) из периферической крови в мозг.
Кроме того, инсулин оказывает регуляторное действие на экспрессию и активность некоторых транспортных белков. Так, инсулин повышает экспрессию гликопротеина-Р (170-кДа), играющего важную роль в поддержании целостности ГЭБ (38), и подавляет экспрессию и активность белка лекарственной устойчивости рака молочной железы (39). Нейрохимические изменения в капиллярах мозга в ответ на действие инсулина реализуются через снижение активности щелочной фосфатазы (40). Вместе с тем наблюдается активация элемента антиоксидантного ответа 4 и увеличение экспрессии каталитической субъединицы глутаматцистеин лигазы (41). Инсулин-зависимое снижение активности серотониновых рецепторов подсемейства 5-HT2c в сосудистом сплетении мозга указывает на роль MAP-киназного пути в реализации эффекта серотонинового рецептора (42).
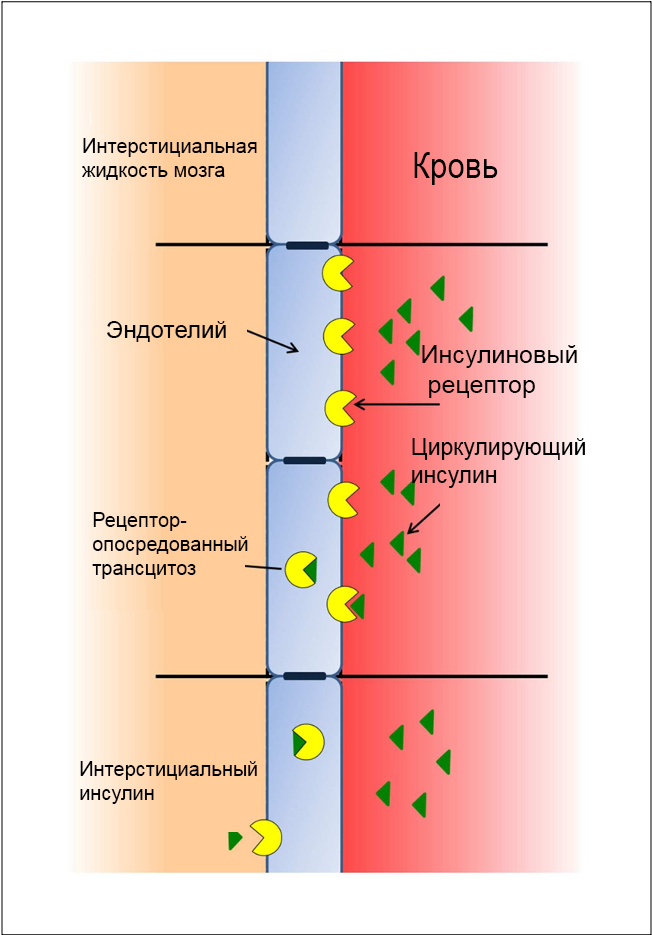
Рисунок 1. Проникновение инсулина через ГЭБ опосредовано действием белков-переносчиков
Ферменты, участвующие в деградации инсулина, также в большом количестве присутствуют в нейронах коры больших полушарий, гиппокампа, мозжечка и ствола, а также в олигодендроцитах, сосудистом сплетении и эндотелиальных клетках (43). При низком уровне пептида Aβ наблюдается увеличение активности ферментов, разрушающих инсулин, что указывает на их роль в катаболизме Aβ и может стать терапевтической мишенью при разработке подходов к лечению нейродегенеративных заболеваний (44).
Механизмы действия инсулина в ЦНС
Инсулиновые рецепторы в мозге
Ген ИР расположен на хромосоме 19p13.2–19p13.3 и содержит 22 экзона, 11 из которых кодируют α и β субъединицы белка. В результате альтернативного сплайсинга +/− экзона 11 образуются две изоформы белка-предшественника – ИР-B и ИР-A, соответственно. Этот экзон кодирует небольшую последовательность аминокислот, расположенных на С-терминальном конце внеклеточного домена α-субъединицы рецептора (45). ИР-B содержатся преимущественно в инсулин-чувствительных тканях человека – скелетных мышцах, адипоцитах и печени, в то время как рецепторы типа А преобладают в структурах ЦНС (46–48).
Гетеротетрамер ИР содержит два лиганд-связывающих сайта – внеклеточные гидрофильные α-субъединицы (15 сайтов N-гликозилирования и 37 цистеиновых остатков) и две трансмембранные β-субъединицы, связанные друг с другом дисульфидными связями. β-субъединица инсулинового рецептора обладает тирозинкиназной активностью (49).
Началу активного изучения ИР в мозге в первой половине 1970-х годов предшествовало исследование на крысах, в котором было показано снижение уровня глюкозы в крови в ответ на введение 500 μU инсулина в сонную артерию (50). Кроме того, в других исследованиях на животных было показано активное связывание меченого 125I-инсулина на мембранах клеток различных тканей (51).
Метаболизм углеводов в печени регулируется преимущественно посредством холинергической активации, нежели изменением секреторной активности островковых клеток поджелудочной железы. Впервые ИР были обнаружены в структурах мозга в 1978 году (52) и присутствовали на всех исследуемых этапах развития (53). С этих пор широкое распространение ИР в ЦНС было неоднократно подтверждено in vitro и in vivo.
Использование радиоактивной метки для оценки содержания инсулина в культурах тканей показало преобладание ИР на поверхности клеток передней части гипоталамуса и значительно более низкое количество ИР на мембранах клеток задней доли гипоталамуса, таламуса и коры больших полушарий (54). Активное связывание меченого инсулина [125I] также было показано в структурах обонятельной и лимбической систем в новой коре и корковых областях, получающих афферентные сигналы от базальных ганглиев, гиппокампе, мозжечке и сосудистом сплетении мозга, что подтверждает нейромодуляторную роль инсулина в ЦНС (55). Методами авторадиографии и компьютерной денситометрии показано преобладание ИР в зонах, связанных с обонянием, пищевым поведением и автономными функциями (56). Данные гибридизации in situ демонстрируют высокий уровень мРНК ИР в клетках гранулярного слоя обонятельной луковицы, мозжечка и зубчатой извилины, а также пирамидных клетках пириформной коры, гиппокампа, сосудистого сплетения и дугообразного ядра гипоталамуса (57).
Экспрессия мРНК ИР выше у крыс Цукера с ожирением (fa/fa) по сравнению с крысами с нормальной массой тела (Fa/−) (58). Однако связывание инсулина в культуре клеток мозга нормальных крыс не отличается от связывания гормона у крыс с стрептозоцин-индуцированным диабетом, что опровергает данные о повышении экспрессии рецепторов инсулина при СД (59).
Наряду с IR, рецепторы инсулиноподобного фактора роста (ИФР1-Р) также широко распространены в ЦНС, в частности в обонятельных и сенсорных зонах мозга и областях, отвечающих за контроль автономной регуляции, включая гипофиз (60). Кроме того, характер экспрессии ИР и ИФР1-Р в мозге крыс обладает межполушарной асимметрией и различается у самцов и самок. Предполагается, что различия в пространственном распределении этих рецепторов могут лежать в основе патогенеза некоторых психических расстройств и поведенческих различий, в частности связанных с активностью гиппокампа (например, пространственное обучение и адаптивный ответ на стресс), между мужчинами и женщинами (61).
Присутствие ИР в гипоталамусе, коре больших полушарий и мозжечке в отсутствии СД подтверждено в исследованиях post-mortem (62). Динамика связывания меченого инсулина на мембранах синаптосом коры больших полушарий меняется в процессе развития. Активное связывание инсулина с ИР в мозге обнаруживается уже на 14-й неделе внутриутробного развития, постепенно снижается к 30-й неделе и достигает минимальных значений после рождения (63).
Несмотря на различия в размере (α-субъединица ИР в мозге, ИР-А, меньше по молекулярному весу, чем α-субъединица периферического ИР, ИР-В), степени гликозилирования (выше в периферических ИР) и специфичности, кинетика и фармакологические свойства ИР в ЦНС не отличаются от свойств инсулиновых рецепторов в периферических тканях (64). С другой стороны, связывание инсулина центральными и периферическими ИР запускает разные молекулярные каскады. Так, избыток инсулина в периферических тканях вызывает снижение экспрессии ИР, и не влияет на экспрессию ИР в мозге (65). Такая гетерогенность действия центральных и периферических ИР способствует независимой и специфичной регуляции клеточных каскадов в ответ на связывание одних и тех же лигандов.
Активность экспрессии ИР также определяется типом рецептора (66). Плотность ИР на мембранах нейронов меняется на каждом этапе развития мозга. В процессе активного нейрогенеза наиболее высокий уровень ИР обнаружен в таламусе, хвостатом ядре и скорлупе, а также некоторых ядрах среднего мозга и ствола. В мозге взрослых особей плотность ИР в этих структурах значительно снижается (67).
Система инсулиновой сигнализации в мозге
Инсулиновый рецептор принадлежит семейству рецепторных тирозинкиназ. Связывание инсулина с α-субъединицей ИР в нейронах и клетках глии стимулирует фосфорилирование и активацию β-субъединицы, обладающей тирозинкиназной активностью (68). У млекопитающих механизм инсулиновой сигнализации (Рисунок 2) регулируется посредством фосфорилирования тирозиновых остатков в составе некоторых белков, включая субстраты ИР (ИРС) (69) и адапторные белки (70), которые способствуют интеграции действия рецепторных тирозинкиназ и других звеньев сигнального пути (71). Связывание инсулина вызывает интернализацию ИР в составе окаймленных везикул (72). Этот процесс играет основополагающую роль в инсулиновой сигнализации. Попадая внутрь клетки, ИР деградируют под действием внутриклеточных ферментных комплексов или снова встраиваются в мембрану.
Рисунок 2. Трансдукция сигнала и реализация физиологических эффектов инсулина и ИФР-1
Действие инсулина в ЦНС реализуется преимущественно за счет ИРС-1 и ИРС-2. ИРС-1 участвует в регуляции роста и опосредует периферическое действия инсулина, в то время как ИРС-2 играет важную роль в созревании нейронных структур, контроле массы тела, гомеостазе глюкозы и репродуктивной функции самок (74). На NH2-терминальном конце ИРС белков содержится домен гомологии плекстрина (РН), за которым следует фосфотирозин-связывающий (PTB) домен. На С-терминальном конце находятся сайты фосфорилирования тирозина и серина/треонина (75).
Сайты фосфорилирования тирозина участвуют в координации сигнальных каскадов посредством связывания SH2 доменов эффекторных (фосфоинозитид-3-киназа, PI3K; фосфатаза SHP2; тирозинкиназа Fyn) или адапторных белков (SOCS1, SOCS-3, GRB2 и др.) (70, 74). Фосфорилирование серина ИРС-1/2 с-Jun N-терминальной киназой (JNK1) и другими протеинкиназами, наоборот, подавляет фосфорилирование тирозина в ответ на связывание инсулина. Это, наряду с убиквитин-опосредованной деградацией ИРС-1/2, играет важную роль в развитии инсулинорезистентности (76, 77). С другой стороны, повышение экспрессии ИРС-2 в ответ на действие цАМФ и активации CREB способствуют усилению инсулиновой сигнализации (78).
Синапс образует физический контакт между нейронами и способствует передаче сигналов между клетками. На роль ИРС сигнализации в постнаптическом аппарате нейронов указывают данные о коэкспрессии ИР и тирозинкиназного субстрата ИР p58/53 (IRSp53) в богатых синапсами молекулярном и гранулярном слоях мозжечка, а также в синапсах изолированных клеток гиппокампа (79). Фосфорилирование IRSp53 в ответ на действе инсулина (80, 81) играет важную роль в реорганизации цитоскелета, предшествующей росту нейритов (82), а также участвует в патогенезе ряда нейродегенеративных заболеваний (83). Это подтверждается результатами исследований на животных, демонстрирующих, что нокаут гена IRSp53 вызывает нарушения когнитивных функций, выявляемых в лабиринте Морриса и тесте на распознавание новых объектов (84).
Взаимодействие ИРС и PI3K приводит к активации PI3K и последующему фосфорилированию компонента плазматической мембраны инозитола PI (4,5)P2 с образованием инозитолтрифосфата PI (3,4,5)P3. Это, в свою очередь, стимулирует ассоциацию серинтреониновой PDK киназы (3-фосфоинозитол-зависимая протеинкиназа) и протеинкиназы В (PKB или Akt) с плазматической мембраной клетки, а также активацию Akt в ответ на фосфорилирование киназами PDK1 и PDK2 (85). Этот сигнальный путь подавляется действием липидной фосфатазы PTEN или SHIP2. Фосфорилирование TSC2 (комплекс туберозного склероза 2, туберин 2) киназой Akt приводит к активации мишени рапамицина млекопитающих (mTOR) – цитоплазматической протеинкиназы, играющей ключевую роль в регуляции клеточной пролиферации и метаболизма, и таким образом опосредует связь между инсулиновой сигнализацией и чувствительностью к уровню нутриентов (70, 86). Помимо IRS/PI3K/Akt сигнального пути существует периферический путь инсулиновой сигнализации, способствующий транслокации транспортера глюкозы GLUT-4 в ответ на действие инсулина и включающий ряд молекулярных субстратов ИР – Cbl и APS. Интеграция различных белков в состав липидных рафтов индуцирует слияние везикул, содержащих GLUT-4, с плазматической мембраной клетки (71, 85).
Связывание инсулина с ИР также приводит к фосфорилированию остатков тирозина в адапторных белках Gab-1/Shp2, Shc/Grb2 и SOS/Grb2, активации G-белка Ras и запуску сигнального каскада митоген-активируемой протеинкиназы (МАР), в частности MAPK/ERK киназы (MEK) и киназы, регулируемой внеклеточными сигналами (88). МАР-киназа ERK (extracellular signal-regulated kinase) активирует ряд цитоплазматических белков, включая S6 рибосомальную киназу p90rsk (89), белки цитоскелета, фосфолипазу А2 (PLA2), а также сигнальные белки – рецепторные тирозинкиназы, рецепторы эстрогена и белки семейства SOS и STAT (сигнальный белок и активатор транскрипции). Попадая в ядро, ERK фосфорилирует факторы транскрипции – белки семейства Ets – и участвует в контроле экспрессии генов (18, 70).
Нарушение экспрессии и функциональной активности ИР в процессе развития, в том числе в результате точечных мутаций F382V (нарушение транспорта компонентов ИР к поверхности клетки), R735S (инсулинорезистентность в результате ингибирования синтеза белков-предшественников), L1018A (подавление тирозинкиназной активности рецептора) и Y960F (различные функциональные нарушения) лежит в основе патогенеза некоторых функциональных нарушений мозга (49).
Действие инсулина в ЦНС
Роль инсулина в регуляции энергетического обмена, гомеостаза глюкозы и пищевого поведения
Глюкоза – основной источник энергии для клеток мозга вне условий голодания, когда источником служат кетоновые тела (90). Наряду с метаболической, глюкоза выполняет сигнальную функцию и участвует в регуляции углеводного гомеостаза в ЦНС и периферических тканях.
Направленность действия глюкозы в ЦНС определяется присутствием двух типов рецепторов на поверхности чувствительных нейронов, играющих важную роль в регуляции пищевого поведения, энергетического обмена и углеводного гомеостаза (49). Связывание глюкозы рецепторами первого типа (glucose-excited, GE) вызывает активацию клеток, в то время как действие глюкозы на рецепторы второго типа (glucose-inhibited, GI) подавляет их активность. Сенсором глюкозы в чувствительных нейронах служит глюкокиназа, которая участвует в поддержании энергетического баланса и контроле потребления пищи (91–94).
Эффективность захвата глюкозы определяется уровнем экспрессии белков транспортеров (Таблица 1) и сенсоров глюкозы (95). Транспортный белок GLUT-1 наиболее широко представлен в ЦНС. Две изоформы белка-транспортера различаются степенью гликозилирования. В астроцитах экспрессируется изоформа с молекулярным весом 45 кДа, устойчивая и гипо- и гипергликемии; экспрессия изоформы с молекулярным весом 55 кДа встречается преимущественно в эндотелии, повышается в ответ на гипогликемию и устойчива к гипергликемии.
Функция GLUT-1 в структурах ЦНС определяется специализацией клеток, поэтому инсулин может оказывать разнонаправленное действие на активность транспортных белков (49, 96). GLUT-2, совместно с глюкокиназой и рецептором сульфонилмочевины 1 (SUR1), экспрессируется преимущественно в латеральной области гипоталамуса и некоторых его ядрах – паравентрикулярном и дугообразном (97, 98, 49, 93, 99). В нейронах мозжечка, стриатума, коры больших полушарий и гиппокампа, а также в некоторых глиальных клетках и эндотелии преобладают транспортеры глюкозы типа GLUT-3, активность которых повышается при низком уровне глюкозы.
Таблица 1. Основные изоформы транспортеров инсулина в мозге
| Изоформы GLUT | Расположение | Тип клеток | Распространенность | Регуляция |
| GLUT-1 | Повсеместно | Глиальные клетки и эндотелий сосудов | Присутствуют в большом количестве | Гипогликемия, инсулин |
| GLUT-2 | Гипоталамус | Нейроны, глиальные клетки, танициты | Присутствуют в ограниченном количестве |
|
| GLUT-3 | Мозжечок, стриатум, кора и гиппокамп | Нейроны, глиальные клетки и клетки эндотелия | Присутствуют в большом количестве |
|
| GLUT-4 | Обонятельная луковица, зубчатая извилина гиппокампа, гипоталамус и мозжечок | Нейроны, глиальные клетки | Присутствуют в большом количестве в некоторых областяхr | Глюкоза, инсулин, физическая активность |
| GLUT-8 | Гипоталамус, мозжечок, ствол мозга, гиппокамп, зубчатая извилина, первичная обонятельная кора | Тела нейронов и апикальные дендриты | Присутствуют в ограниченном количестве | Глюкоза |
В отличие от периферических тканей, мозг долгое время считался нечувствительным к инсулину, в том числе из-за низкого уровня транспортеров GLUT-4 в структурах ЦНС. Наиболее высокий уровень GLUT-4 обнаружен на мембране и в цитоплазматическом пуле клеток обонятельной луковицы, зубчатой извилины гиппокампа, гипоталамуса и коры больших полушарий, хотя в этих структурах он не превышает содержание GLUT-1 и GLUT-3 (102). Наиболее высокий уровень GLUT-4 обнаружен в мозжечке, где его экспрессия напрямую регулируется действием инсулина (103). В ответ на внутривенное введение глюкозы и увеличение уровня инсулина повышается встраивание цитоплазматических форм GLUT-4 в мембрану клеток мозжечка, коры и гиппокампа (104). В GE и GI нейронах гипоталамуса GLUT-4 коэкспрессируется с ИР и глюкокиназой, что способствует повышению захвата глюкозы в ответ на действие инсулина (105).
Однако инсулин-зависимая регуляция не является доминирующим механизмом контроля транспорта глюкозы. Это подтверждается данными о наличии нейронального ответа на изменение уровня глюкозы даже в отсутствии инулина (97, 98, 106). Кроме того, показано, что инсулин не влияет на захват глюкозы в гиппокампе, а связывание инсулина с ИР не вызывает увеличения AS160-зависимой транслокации GLUT-4 (104).
Транспортер GLUT-8 обнаруживается только в структурах ЦНС, в частности в возбуждающих и тормозящих нейронах гиппокампа, и при нормальных физиологических условиях и в экспериментальной модели СД1 (107) экспрессируется только на мембране тел нейронов и проксимальных участках апикальных дендритов (108, 109). Хотя физиологическая роль GLUT-8 до сих пор малоизучена, чувствительность GLUT-8 к инсулину указывает на роль этого транспортера в мобилизации субстрата в условиях дефицита глюкозы (110).
Внутри клетки GLUT-8 способствует транспорту глюкозы из шероховатого эндоплазматического ретикулума (ЭР) в цитоплазму и поддержанию гомеостаза глюкозы в клетках гиппокампа, обладающих высокой чувствительностью к гипергликемии и снижению уровня инсулина (111, 112). Введение глюкозы вызывает активацию GLUT-8 и увеличение транспорта глюкозы из цитоплазмы в шероховатый ЭР, но не стимулирует встраивание белка в мембрану.
Подавление действия инсулина в гипоталамусе или связывание инсулина с нейронами дугообразного ядра снижает регуляторное влияние инсулина на синтез глюкозы в печени (113) и может вызывать нарушение контроля глюконеогенеза (114) и гипергликемию у пациентов с СД (115, 116).
Действие инсулина на активность нейронов гипоталамуса опосредовано АТФ-чувствительными K+-каналами (117), активация которых вызывает гиперполяризацию и снижение ответа на глюкозу (118). Резекция отростков блуждающего нерва, через которые клетки печени получают афферентацию от гипоталамуса, также подавляет ингибирующее действие инсулина на образование глюкозы в гепатоцитах (116). Эти данные подтверждают результаты работы Claude Bernard (119), который в 1855 году показал, что разрушение четвертого желудочка вызывает глюкозурию у мышей.
Популяции нейронов, участвующих в поддержании энергетического гомеостаза, расположены преимущественно в гипоталамических центрах голода и насыщения. Как и GLUT-2, глюкокиназа (92–94), AMPK и PASK и рецепторы орексигенных и анорексигенных молекул, которые продуцируются клетками этих ядер, выступают в качестве сенсоров энергетического обмена, генерируя интегрированный ответ на афферентную стимуляцию при изменениях энергетического гомеостаза или дефиците ресурсов. Гипоталамические центры голода и насыщения детектируют сигналы об уровне глюкозы и передают их в другие зоны мозга, что приводит к активации пищевого поведения. Активация PI3K каскада является общим звеном сигнального каскада инсулина, лептина и серотонина, и играет важную роль в патогенезе инсулинорезистентности и контроле потребления пищи. В периферических тканях инсулин стимулирует анаболические процессы, в то время как в структурах ЦНС он проявляет анорексигенные свойства и выполняет катаболическую роль (120). Кроме того, в гипоталамусе инсулин активирует JAK2 и SHT3 и усиливает действие лептина (121). Механизмы резистентности нейронов гипоталамуса к инсулину и лептину у мышей с СД2 открывают новые возможности в изучении механизмов инсулинорезистентности при СД2 (33).
Роль инсулина в регуляции полового поведения
Репродуктивная способность во многом зависит от энергетического метаболизма, и нарушение гомеостаза глюкозы может нарушать нормальную регуляцию полового поведения (122), изменяя активность гипоталамо-гипофизарно-гонадальной оси (123). Достаточный уровень энергетических ресурсов способствует нормальному функционированию репродуктивной системы и повышает выживаемость потомства.
Воздействие низких концентраций инсулина на клетки гипоталамуса стимулирует секрецию рилизинг-фактора лютеинизирующего гормона (ЛГ-РГ) и зависит от гомеостаза глюкозы (124), что подтверждает его роль в регуляции репродуктивной функции. С другой стороны, воздействие инсулина в высокой концентрации не влияет на секрецию ЛГ-РГ (124). Введение инсулина в культуру клеток коры больших полушарий также повышает частоту импульсной секреции лютеинизирующего гормона (ЛГ), в то время как при введении глюкозы такой эффект не наблюдается (125). У мышей с СД низкий уровень инсулина в ЦНС вызывает снижение высвобождения ЛГ (124), которое восстанавливается при введении инсулина в мозг или периферические ткани (126). В других исследованиях показано, что низкий уровень инсулина у крыс с СД способствует снижению секреции гонадотропин-рилизинг гормона (ГнРГ) в гипоталамусе и подавлению ответа гонадотропных клеток гипофиза на действие ГнРГ (126). Таким образом, инсулин играет важную роль в контроле импульсной секреции ГнРГ (127). Однако авторы отмечают, что секреция ЛГ зависит не только от действия инсулина, но и от содержания глюкозы. Это опосредовано детекторами глюкозы в гипоталамусе, которые также могут влиять на секрецию ГнРГ, независимо от уровня инсулина (128).
Влияние инсулина на клеточную пролиферацию и дифференцировку
Трофическая функция инсулина в ЦНС реализуется через регуляцию клеточной пролиферации, дифференцировки и роста нейритов в процессе эмбрионального развития. Внутривенное введение инсулина повышает активность орнитин декарбоксилазы в мозге новорожденных крыс, которая служит маркером нейронального развития (129). Участие инсулина в процессах пролиферации подтверждается данными о повышении количества ИР в период активной клеточной дифференцировки (130). Действие ИР на созревание (131) мозга, и также рост и регенерацию аксонов (132) реализуется посредством ИРС-2.
Нейротрофическая функция инсулина была продемонстрирована в исследованиях in vitro. В мозге крыс инсулин стимулирует пролиферацию (133), а в культуре нейронов переднего мозга курицы активирует рост и дифференцировку нейронов (134). ИР, участвующие в реализации эффекта инсулина на нейрональный рост и развитие, также найдены на мембранах глиальных клеток (135, 136), в которых направленность их действия определяется типом клеток. Кроме того, инсулин и ИФР2 стимулируют активность фактора роста нервов (ФРН), необходимого для роста нейритов (137). Действие инсулина на развитие ЦНС зависит от присутствия астроцитов (138), в которых наблюдается активная пролиферация белков сигнального каскада инсулина (139, 140).
В культуре эмбриональных клеток инсулин повышает фосфорилирование рибосомального белка S6 (136) и активность протеинкиназы-эпсилон в цитоплазме клетки (141), стимулируя рост нейритов (142, 143).
Механизм действия инсулина на нейрональное развитие также включает другие ферменты, в том числе фосфатидилинозитол-3-киназу (PI3K) (144). Активация сигнального пути PI3K/mTOR в ответ на действие инсулина повышает экспрессию белка постсинаптической плотности PSD-95 в нейронах CA1 и способствует формированию дендритных шипиков и возбуждающих синапсов в клетках гиппокампа (145, 146).
В инсулин-зависимой регуляции роста нейритов также участвует тау-белок, ассоциированный с микротрубочками. Эффект тау-белка на аксональный рост реализуется через PI3K/mTOR каскад и сопровождается повышением экспрессии мРНК и уровня тубулина (147).
Эндогенный инсулин способствует формированию нейрофиламентов (148) и участвует в регуляции пролиферации и дифференцировки плюрипотентных стволовых клеток. Недостаточность инсулина вызывает неапоптотическую клеточную гибель (130), а снижение активности PI3K/Akt каскада нарушает процесс дифференцировки стволовых клеток человека (hNSC), которые, в отличие от стволовых клеток грызунов, обладают высокой чувствительностью к инсулину и функционируют в узком диапазоне концентрации гормона (149). Таким образом, инсулин играет ключевую роль в интеграции нейрональной активности и регуляции энергетического гомеостаза, определяющих каждый этап клеточной дифференцировки (150).
Нейропротективный эффект инсулина
Нейропротективный эффект инсулина проявляется в подавлении апоптоза, инактивации β-амилоидного белка, снижении оксидативного центра и ишемии. Важную роль в реализации антиапоптотического эффекта инсулина играет активность PI3K/Akt/mTOR пути и белка p70SK. Ингибирование mTOR рампамицином снижает антиапоптотический эффект инсулина, что указывает на важную роль PI3K/Akt/mTOR пути и p70SK белка в реализации этого эффекта (151). Кроме того, инсулин снижает образование Aβ фибрилл, предотвращая клеточную гибель в результате накопления β-амилоида (152).
Образование активных форм кислорода (АФК) в результате оксидативного стресса индуцирует окисление липидов и белков, вызывая нарушения функциональной активности GLUT-3 и захвата глюкозы. Это приводит к накоплению лактата, ацидозу и митохондриальной дисфункции (153). В условиях оксидативного стресса инсулин стимулирует поглощение глюкозы и образование пирувата, способствуя восстановлению внутриклеточной концентрации АТФ (154). Кроме того, инсулин предотвращает внесинаптическое накопление глутамата и гама-аминомасляной кислоты (ГАМК) в результате снижения захвата этих нейромедиаторов при оксидативном стрессе (155) и стимулирует накопление мочевой кислоты, обладающей антиоксидантной активностью (156).
Антиишемическое действие инсулина реализуется с помощью двух основных механизмов – прямого действия на нейрональные рецепторы и повышения уровня глюкозы в периферических тканях (157). При транзиторной ишемии инсулин повышает уровень ГАМК – главного тормозного нейромедиатора в мозге – во внеклеточном пространстве (158).
Стимуляция Na+/K+ АТФазы в ответ на действие инсулина приводит к снижению внеклеточной концентрации K+ и внутриклеточной концентрации Na+, способствуя уменьшению нейрональной активности и метаболических потребностей клеток, предотвращению накопления жидкости и образованию постишемического отека тканей, а также снижению уровня лактата (159, 160).
С другой стороны, реперфузия при ишемии мозга стимулирует фосфорилирование JNK1/2, экспрессию Bcl-2 и деградацию каспазы-3 в гиппокампе крыс, что указывает на важность интегрированной активности Akt и JNK1/2 в реализации противоишемического действия инсулина.
Нейромодуляторный эффект инсулина
Нейромодуляторное действие инсулина реализуется через регуляцию активности ионных токов и контроль уровня и действия нейромедиаторов. Электрофизиологический эффект инсулина in vivo опосредован действием ГАМК, которое снижается в ответ на введение ингибиторов ИР (161). Кроме того, рецепторы ГАМК являются одним из основных субстратов Akt фосфорилирования, что подтверждает роль инсулина в регуляции плотности рецепторов ГАМК на постсинаптической мембране (162).
Изменение активности ионных каналов в ответ на действие инсулина способствует реорганизации ионных токов и изменению активности нейронов. В нейронах гипоталамуса инсулин повышает активацию K+АТФ каналов, что приводит к гиперполяризации мембраны и торможению нейрональной активности (163). Кроме того, инсулин стимулирует Na+/K+ АТФазу и вызывает увеличение внутриклеточной концентрации Ca2+, стимулируя высвобождение нейропептидов (164).
Инсулин способствует подавлению высвобождения норадреналина и повышению захвата серотонина в нейронах (165, 166), и, как следствие, увеличению эффективности метаболизма глюкозы (165, 166). В нейронах стриатума крыс с стрептозоцин-индуцированным СД (167) и при введении галоперидола (168), инсулин увеличивает плотность дофаминовых рецепторов, хотя у здоровых животных этот эффект не наблюдается. Системное введение инсулина вызывает повышение уровня дофамина и серотонина в ЦСЖ и снижение плотности
α2-адренергических рецепторов в нейронах гипоталамуса (169). Помимо этого, инсулин стимулирует захват аминокислот, необходимых для синтеза нейромедиаторов (170).
Влияние инсулина на когнитивные функции и память
Внутривенное введение, внутрицеребровентрикулярная или внутригиппокампальная инфузия инсулина связаны с улучшением когнитивных функций и памяти (171), которые сопровождаются увеличением экспрессии ИР и активацией сигнальных каскадов в гиппокампе (172). Инсулин также способствует восстановлению памяти при ишемическом поражении мозга (173).
Введение низких доз стрептозоцина в мозг вызывает центральную инсулинорезистентность подавляет активность инсулина и приводит к нарушениям памяти и поведения (174). СД1 и СД2 повышают риск когнитивных нарушений и развития БА у пожилых пациентов. В эпидемиологических исследования показано, что введение инсулина больным с СД1/СД2 и БА улучшает формирование памяти и способствует поддержанию уровня глюкозы (175). Системное введение инсулина здоровым испытуемым в условиях эугликемии и гиперинсулинемии улучшает показатели вербальной памяти и селективного внимания.
Ключевую роль в формировании кратковременной и долговременной памяти играют процессы долговременной потенциации (ДП) и долговременной депрессии (ДД) (176). ДП возникает в ответ на длительную и синхронную активацию пресинаптических и постсинаптических нейронов, способствующую поддержанию деполяризации постсинаптической мембраны в результате повышения уровня Ca2+ и консолидации. ДД является компенсаторным механизмом и вызывает снижение эффективности нейрональной передачи в ответ на интенсивное воздействие стимула. Наряду с ДП и ДД, важную роль в процессах обучения и памяти играет пластичность дендритных шипиков и реорганизация цитоскелета, опосредованные действием глутамата на AMPA и NMDA рецепторы. Активность рецепторов регулируется изменением их плотности на клеточной мембране или ковалентной модификацией белковых субъединиц. В процессе ДП наблюдается увеличение плотности AMPA рецепторов на постсинаптической мембране, в то время как ДД приводит к снижению количества рецепторов. Фосфорилирование и дефосфорилирование глутаматных рецепторов при ДП и ДД играет важную роль в контроле эффективности нейронной передачи (177).
Инсулин вызывает снижение плотности AMPA рецепторов на постсинаптической мембране, способствуя ДД. Этот процесс во многом зависит от уровня фосфорилирования ИР, активности PI3К и синтеза белка (178). Кроме того, инсулин стимулирует фосфорилирование субъединицы GluR2 AMPA рецепторов в нейронах гиппокампа, способствуя их интернализации и снижению возбудимости клеток (179).
Важную роль в реализации действия инсулина на процессы обучения и памяти играет ГАМК. Инсулин стимулирует транслокацию и экспрессию рецепторов ГАМК на постсинаптической мембране, в то время как подавление активности PI3K снижает этот эффект (180).
Стимулирующее действие ИФР1 на активность нейронов в гиппокампе крыс реализуется с участием AMPA рецепторов и PI3К (181). О роли соматотропного гормона (СГ) в ДП и формировании памяти свидетельствует повышение экспрессии NMDA рецепторов в гиппокампе (182).
Воспаление и инсулинорезистентность
У пациентов с ожирением и СД2 воспаление является ключевым звеном патогенеза инсулинорезистентности в ЦНС и периферических тканях и повышает риск развития когнитивных нарушений и БА (183). У пациентов с БА обнаружено увеличение уровня интерлейкина IL-6 в ЦСЖ (184), что указывает на роль воспаления в аккумуляции β-амилоидного пептида Aβ (185). Результаты экспериментальных исследований демонстрируют противовоспалительное действие инсулина у пациентов с БА (186).
Введение липополисахарида приводит к увеличению уровня С-реактивного белка и провоспалительных цитокинов IL-1β, IL-6 и TNF в плазме крови (187). TNFα и IL-6, в свою очередь, стимулируют активацию NFkβ и транскрипцию провоспалительных факторов TNFα, IL-6 и IL-1b (188). Это приводит к нарушению синаптической пластичности в гиппокампе и формированию пространственной памяти (189).
Инсулинорезистентность периферических тканей у пациентов с ожирением связана с увеличением уровня провоспалительных цитокинов и свободных жирных кислот. Хроническое воспаление также способствует развитию инсулинорезистености, СД2 и БА (190). В частности, повышение уровня TNFα и Aβ в мозге пациентов с гиперинсулинемией стимулирует образование амилоидных бляшек (191). Среди пациентов с ожирением и БА наблюдается снижение концентрации инсулина в мозге, что указывает на нарушение транспорта инсулина через ГЭБ и снижение чувствительности нейронов к действию гормона (192).
В зависимости от типа рецепторов – TNF-R1 или TNF-R2 – TNF-α проявляет нейротоксический или нейропротективный эффект. TNF-R1 оказывает проапоптотическое действие, в то время как TNF-R2 предотвращает клеточную гибель. У пациентов с БА, диабетом и нарушением толерантности к глюкозе наблюдается увеличение уровня TNF-R1 и снижение уровня TNF-R2 (193, 194), который нормализуется после трех недель высококалорийной диеты (195). Накопление конечных продуктов гликирования, оксидативный стресс и повреждение клеточных структур способствуют нарушению когнитивных функций у пациентов с СД (196).
Нарушение каскадов инсулиновой сигнализации, в частности PI3K/Akt и киназы-3 гликогенсинтетазы GSK-3, связано с повышением воспаления и инсулинорезистентностью (197). Известно, что PI3K подавляет образование IL-12 в дендритных клетках, а GSK-3 способствует гиперфосфорилированию и регуляции метаболизма Aβ (198).
Активация ИР приводит к увеличению фосфорилирования и подавлению активности GSK-3β (199). У пациентов с БА и СД2 наблюдается повышение активности GSK-3β и увеличение фосфорилирования ИР и ИРС-1 (200). Кроме того, сигнальный каскад PI3K/Akt/GSK-3 играет важную роль в активации STAT-3 в глиальных клетках (201): ингибирование GSK-3 вызывает повышение уровня противовоспалительных цитокинов, в частности IL-10, и снижение образования провоспалительных факторов – IL-1β, IL-6 и IFN-γ (202).
Известно, что воспаление играет ключевую роль в патогенезе БА, способствуя повышению продукции провоспалительных факторов Т-лимфоцитами, моноцитами и клетками глии (203, 204). Данные исследований на животных показывают, что подавление продукции провоспалительных цитокинов позволяет снизить выраженность симптомов инсулинорезистентности (205). Хотя воспаление не участвует в развитии острой инсулинорезистентности, которая развивается в ответ на рацион с высоким содержанием жира (206), оно оказывает значительное влияние на развитие хронической инсулинорезистентности (207).
Роль TNF-α в патогенезе инсулинорезистентности реализуется через подавление тирозинкиназной активности ИР и снижение активности SOCS-3, приводя к инактивации ИРС-1 (208). Кроме того, активация JNK в ответ на повышение уровня факторов воспаления и свободных жирных кислот повышает фосфорилирование ИРС-1 и снижает эффективность инсулиновой сигнализации, способствуя увеличению уровня воспаления и развитию инсулинорезистентности. Как показывают исследования на животных, инактивация JNK и подавление воспалительного ответа позволяют избежать негативных метаболических эффектов, вызванных гиперлипидемией (210, 211, 212).
Внутрирецебровентрикулярная инфузия TNF-α стимулирует воспаление в гипоталамусе, повышение уровня инсулина и нарушение инсулиновой сигнализации в периферических тканях. Считается, что центральная инсулинорезистентность служит адаптивным механизмом при высококалорийной диете и сопровождается нарушением гомеостаза глюкозы. Повышение уровня свободных жирных кислот служит сигналом для активации высвобождения провоспалительных цитокинов, подавляющих действие инсулина (213).
У пациентов с БА показано нарушение инсулиновой сигнализации, снижение тирозинкиназной активности ИР, подавление экспрессии мРНК инсулина и ИФР1 и их рецепторов (214). Это вызывает снижение эффективности сигнальных каскадов инсулина (IR/IRS-1/PI3K) и ИРФ1 (IGF1R/IRS-2/PI3K). Уменьшение ответа на действие инсулина опосредовано фосфорилированием остатков серина (IRS-1 pS616, IRS-1 pS636/639) и инактивацией ИРС-1. При умеренных и выраженных когнитивных нарушениях наблюдается повышение маркеров инсулинорезистентности, независимо от уровня аполипопротеина E (APOE-4) (104). Снижение чувствительности к инсулину также сопровождается повышенным накоплением Aβ, который, в свою очередь, оказывает тормозящее действие на экспрессию и функциональную активность гормона (215). Пептид Aβ проявляет антагонистическое действие по отношению к инсулину, конкурентно связываясь и снижая аутофосфорилирование ИР (216, 217). Это может препятствовать реализации нейропротективного эффекта инсулина у пациентов с БА и СД2 и повышать риск когнитивных нарушений (218). Резистентность к инсулину и ИФР1 служит ранним биомаркером БА и указывает на дисфункцию ИРС-I, вызванную действием Aβ (104).
Сахарный диабет и болезнь Альцгеймера
Результаты последних исследований указывают на ключевую роль инсулина в регуляции нейрональной активности и тесную связь между БА и СД2 (219) (Рисунок 3). БА – самое распространенное нейродегенеративное заболевание, которое сопровождается потерей памяти и прогрессирующей деменцией. В настоящее время более 30 миллионов людей страдают БА, а к 2040 году ожидается рост встречаемости заболевания до 120 миллионов (197).
Важным звеном патогенеза БА является накопление амилоидных бляшек и нейрофибриллярных кубков – агрегатов гиперфосфорилированного тау-белка, а также церебральная амилоидная ангиопатия и нейродегенерация (220).
СД2 сопровождается нарушением секреции инсулина и инсулинорезистентностью. В 2010 году в мире насчитывалось 250 миллионов пациентов с сахарным диабетом, 90% из которых страдали СД2 (221). Наряду с ожирением, старение является одним из ключевых факторов риска БА и СД2.
Рисунок 3. Нарушение инсулиновой сигнализации в патогенезе болезни Альцгеймера. Aβ, β-амилоидный пептид; GLUT-3, транспортер глюкозы 3; GSK-3β, киназа-3 β гликогенсинтетазы; NFT, нейрофибриллярные клубки; PI3K, фосфатидилинозитол-3-киназа.
Инсулинорезистентность, воспаление, накопление амилоидного пептида и когнитивные нарушения являются общими звеньями патогенеза БА и СД2. Наряду с периферической инсулинорезистентностью, в патогенезе этих заболеваний также участвует резистентность к ИФР1 и нарушение функциональной активности ИРС-1 и ИРС-2.
Инсулинорезистентность в периферических тканях предшествует манифестации СД2, что представляет собой важное терапевтическое окно для своевременного лечения (222). Особый интерес вызывает вопрос о роли периферической и центральной инсулинорезистентности в патогенезе БА и СД2. Отсутствие СД2 у пациентов с БА может быть связано с поздней манифестацией симптомов диабета, хотя проявление симптомов когнитивной дисфункции также обнаруживается на поздних стадиях болезни.
Выделают четыре стадии прогрессирования БА. На первой стадии болезнь не сопровождается выраженными симптомами; на преклинической стадии заболевания обнаруживаются патофизиологические изменения на фоне отсутствия выраженных когнитивных нарушений; третья стадия предшествует развитию деменции и сопровождается умеренными когнитивными нарушениями; последняя стадия БА характеризуется тяжелой когнитивной дисфункцией и деменцией (223, 224).
Изменение уровня инсулина и нарушение метаболизма глюкозы повышают риск развития деменции (225, 226). Результаты последних исследований демонстрируют повышение экспрессии маркеров СД2 у пациентов с БА (227) и подтверждают связь между нарушением инсулиновой сигнализации и снижением когнитивных функций. В настоящее время центральная инсулинорезистентность, сопровождающаяся когнитивными нарушениями, рассматривается как отдельная форма СД – сахарный диабет 3 типа (СД3).
Однако эта форма диабета не может считаться классическим проявлением заболевания, так как у пациентов с БА не наблюдается гипергликемии, характерной для СД1 и СД2. Кроме того, в отличие от мышечной и жировой ткани и печени, инсулин не оказывает стимулирующего влияния на захват глюкозы клетками мозга (228). Центральная инсулинорезистентность может развиваться в ответ на изменение гомеостаза глюкозы в периферических тканях и сопровождаться повышением уровня Aβ и транспорта глюкозы (230). Таким образом, данные указывают на центральную роль инсулинорезистентности в манифестации БА и СД2, которой предшествует снижение чувствительности к инсулину в периферических тканях.
Переход от преддиабета к диабету длится от 10 до 15 лет и наиболее широко представлен среди пожилых людей, которые находятся в группе повышенного риска развития БА.
Преддиабет и диабет обнаруживается более чем у 81% пациентов с БА (226). Долгий период развития диабета из преддиабета объясняет, почему инсулинорезистентность у пациентов с БА не всегда сопровождается симптомами СД.
Результаты экспериментальных и эпидемиологических исследований подтверждают связь между инсулинорезистентностью при СД и риском деменции (231). Гиперинсулинемия и гипергликемия, вызванные снижением чувствительности к инсулину, способствуют образованию нейритных бляшек (233). По данным эпидемиологических исследований, с возрастом повышается вероятность коморбидного течения БА и СД, независимо от наличия сердечно-сосудистых заболеваний (234).
В то время как влияние СД2 на активность ЦНС хорошо изучено, существует гораздо меньше данных о центральных эффектах СД1 (235). Рядом исследований показана роль СД1 в нарушении процессов памяти, обучения и когнитивной гибкости (235–242). В основе этого эффекта лежит снижение количества дендритов в сером веществе мозга у пациентов с СД1 (245). Инсулиновая терапия, наоборот, способствует улучшению когнитивных функций (243, 244) и восстановлению морфологической структуры клеток (246). У пациентов с СД1 и СД2 инсулин и ИФР1 предотвращают атрофию головного мозга и когнитивную дисфункцию (247).
Однако существуют данные, опровергающие связь между БА и СД (248, 249). При прогрессирующей форме БА и отсутствии аллеля APOE-4 наблюдается снижение уровня инсулина в ЦСЖ на фоне повышения содежания гормона в крови (197). Увеличение уровня инсулина в плазме крови служит маркером инсулинорезистентности, в то время как снижение уровня инсулина в ЦСЖ указывает на нарушение метаболизма и снижение транспорта инсулина через ГЭБ.
Общими звеньями патогенеза БА и СД являются митохондриальная дисфункция, оксидативный стресс, нарушение метаболизма глюкозы, и образование модифицированных форм ЛПНП (197). Инсулин предотвращает инактивацию окислительного фосфорилирования в митохондриях и обладает протективным эффектом в отношении Aβ и окислительного стресса, в то время как при СД наблюдается снижение активности системы антиоксидантной защиты и повышение чувствительности клеток к действию токсических олигомеров амилоидного белка (250).
Независимо от наличия диабета, нарушение метаболизма глюкозы наблюдаются у большинства пациентов с БА (223), преимущественно в височно-теменной, задней поясной и лобной областях коры. Введение глюкозы стимулирует действие инсулина и способствует улучшению показателей памяти (251, 252).
Недостаточность метаболизма глюкозы связана с нарушением мембранного транспорта, энергетического гомеостаза и метаболизма тиамина. У пациентов с БА наблюдается снижение экспрессии GLUT-1 и GLUT-3 (253), особенно в нейронах коры больших полушарий и зубчатой извилине гиппокампа. Согласно результатам работы Liu et al. (254), снижение метаболизма глюкозы происходит в результате подавления экспрессии транспортных белков и O-GlcN-ацетилирования, повышения фосфорилирования тау-белка и образования нейрофибриллярных бляшек.
БА сопровождается изменением функциональной активности ключевых ферментов цикла Кребса и пентозофосфатного пути: пируватдегидрогеназного комплекса, α- кетоглутаратдегидрогеназного комплекса, транскетолазы и нуклеозид-дифосфатазы (255). Эти данные подтверждают роль митохондриальной дисфункции и изменения метаболизма тиамина в нарушении гомеостаза глюкозы в мозге при БА.
Недостаточность метаболизма глюкозы служит предиктором патофизиологических изменений, ассоциированных с СД (222). Согласно гипотезе Chen и Zhong, нарушение гомеостаза глюкозы и метаболизма тиамина и инсулинорезистентность способствуют агрегации пептида Aβ и гиперфосфорилированию тау-белка, занимая центральное место в патогенезе БА. Эти патофизиологические каскады индуцируют высвобождение провоспалительных факторов, вызывают митохондриальную дисфункцию и оксидативный стресс, повышают уровень конечных продуктов гликирования, апоптоз, эксайтотоксичность и активность протеинкиназ (223).
Общим звеном патогенеза БА и СД также является нарушение метаболизма холестерина. Гиперхолестеринемия вызывает изменение активности островковых клеток поджелудочной железы и снижение секреции инсулина (256, 257). Повышение уровня ЛПНП способствует образованию Aβ и его встраиванию в клеточные мембраны (10). Связывание Aβ с липопротеинами катализирует образование оксихолестерина, обладающего выраженным нейротоксическим действием и способствующего подавлению ERK/Akt каскада (258, 259). Гипергликемия и гиперинсулинемия стимулируют O-GlcN-ацетилирование белков каскада инсулиновой сигнализации и способствуют развитию инсулинорезистентности (260, 261). В образцах лобных областей коры пациентов с СД2 и БА наблюдается снижение активности инсулин/PI3K/Akt сигнального пути и (262) и O-GlcNAc трансферазы, наряду с повышением GSK-3β кальпаин-1-зависимой регуляции, что способствует гиперфосфорилированию белков и нейродегенерации (262).
Образование амилоидных агрегатов, изменение метаболизма Aβ и гиперфосфорилирование тау-белка служат связующим звеном в патогенезе БА и СД. В клетках островков Лангерганса могут формироваться скопления амилоидного белка IAPP, который сходен по структуре с пептидом Aβ и обладает токсическим действием на клетки поджелудочной железы (263, 224). Снижение активности шаперонов, препятствующих агрегации IAPP и Aβ, способствует образованию амилоидных и нейритных бляшек в островковых клетках и нейронах у пациентов с БА и СД (226).
Протеазы играют важную роль в метаболизме и предотвращении накопления продуктов распада Aβ. В частности, металлопептидаза IDE способствует повышению активности инсулина, ИФР и глюкагона. Нарушение инсулиновой сигнализации сопровождается выраженным увеличением уровня фосфорилированного тау-белка и пептида Aβ и способствует клеточной гибели и нейродегенерации, особенно у пациентов с СД2 (264). Повышение уровня инсулина в ЦНС оказывает стимулирующее действие на функциональную активность IDE (265, 266), а также способствует интернализации олигомеров Aβ и предотвращает их связывание с мембраной нейронов.
Важную роль в патогенезе БА играет формирование нейрофибриллярных клубков, которые представляют собой агрегаты гиперфосфорилированного тау-белка (267).
Инсулинорезистентность и периферическая гиперинсулинемия при СД2 приводят к снижению транспорта инсулина через ГЭБ, а системная инсулиновая недостаточность при СД1 повышает фосфорилирование тау-белка (235, 268).
Таким образом, центральная инсулинорезистентность вызывает повреждение нейронов гиппокампа и коры больших полушарий и служит общим звеном патогенеза СД2 и БА. Наряду с нарушением инсулиновой сигнализации, развитию БА могут способствовать вирусные, бактериальные и грибковые инфекции (269, 270, 271). Однако во всех случаях БА сопровождается прогрессирующей нейродегенерацией.
Терапевтические подходы
Понимание механизмов регуляции когнитивных и метаболических функций открывает новые возможности в терапии инсулинорезистентности при СД2 и БА. Ни один из современных терапевтических подходов, направленных на известные звенья патогенеза нейродегенеративных заболеваний – агрегаты амилоидного пептида (272), гиперфосфорилирование тау-белка (273) и метаболизм тиамина, а также использование антиоксидантов и нейропротектиных факторов (274, 275, 276), не позволяет достигнуть высокой эффективности лечения.
В последнее время все большую популярность набирают мультитаргетные подходы, нацеленные на регуляцию молекулярных каскадов, опосредующих патогенетический эффект инсулинорезистентности (276). В частности, наиболее известными молекулярными агентами, способствующими повышению секреции инсулина, являются метформин, антагонисты рецептора, активируемого пролифераторами пероксисом PPARγ, а также миметики инкретина, глюкагоноподобный пептид-1 (ГПП-1) и гастрический ингибирующий полипептид. Наряду с этим показана высокая эффективность интраназального введения инсулина и ГПП-1 в терапии СД2 и при умеренных когнитивных нарушениях (224).
Метформин, представитель класса бигуанидов, способствует поддержанию нормального уровня инсулина натощак и контролирует образование глюкозы в печени. В последнее время появляется все больше данных о способности метформина проникать через ГЭБ, повышая чувствительность к инсулину в структурах ЦНС (277) и способствуя снижению риска деменции у пациентов с СД (278).
Использование агонистов PPARγ, в частности росиглитазона, позволяет добиться значимого улучшения чувствительности к инсулину (279), увеличения функциональной активности адипоцитов, повышения транспорта триглицеридов и жирных кислот из печени и мышц, а также предотвращения накопления Aβ и снижения уровня воспаления (280, 281). Это приводит к нормализации уровня инсулина и улучшению внимания и памяти на ранних этапах патогенеза БА (282). Несмотря на высокую эффективность антагонистов PPARγ, препараты этой группы повышают риск инфаркта миокарда, что ограничивает их применение в терапии инсулинорезистентности.
ГПП-1, представитель класса инкретинов, способствует повышению секреции инсулина в зависимости от уровня глюкозы. В терапии СД широко применяются ингибиторы дипептидилпептидазы (глиптины), агонисты (эксенатид и лираглутид) и миметики ГПП-1. Как и метформин, миметики ГПП-1 способны проникать через ГЭБ и стимулировать соответствующие рецепторы в мозге. Эксенатид и лираглутид оказывают защитное действие на клетки мозга, подавляют нейродегенеративные процессы и предотвращают прогрессирование АД (283, 284, 285). Кроме того, наряду со снижением уровня олигомеров Aβ и нейритных бляшек, агонисты ГПП-1 стимулируют нейрогенез и улучшают распознавание объектов и пространственную память (283, 286, 287) и эффективны в терапии деменции, независимо от наличия СД.
Интраназальное введение инсулина способствует эффективному проникновению гормона в ЦНС и улучшению состояния пациентов с умеренными когнитивными расстройствами и СД2 (240, 288–290, 291). Дополнительной мишенью в терапии инсулинорезистентности служит недостаточность инсулиновой сигнализации в результате фосфорилирования серина и треонина IRS-1 (292).
Список литературы
1. Derakhshan F, Toth C. Insulin and the brain. Curr Diabetes Rev (2013) 9(2):102–16. doi: 10.2174/1573399811309020002
2. Havrankova J, Schmechel D, Roth J, Brownstein M. Identification of insulin in rat brain. Proc Natl Acad Sci U S A(1978) 75(11):5737–41. doi:10.1073/pnas.75.11.5737
3. Dorn A, Bernstein HG, Rinne A, Ziegler M, Hahn HJ, Ansorge S. Insulin and glucagonlike peptides in the brain. Anat Rec (1983) 207(1):69–77. doi:10.1002/ar.1092070108
4. Havrankova J, Roth J, Brownstein MJ. Concentrations of insulin and insulin receptors in the brain are independent of peripheral insulin levels. Studies of obese and streptozotocin-treated rodents. J Clin Invest (1979) 64(2):636–42. doi:10.1172/JCI109504
5. Pardridge WM. Blood-brain barrier biology and methodology. J Neurovirol (1999) 5(6):556–69. doi:10.3109/13550289909021285
6. Banks WA, Jaspan JB, Huang W, Kastin AJ. Transport of insulin across the blood-brain barrier: saturability at euglycemic doses of insulin. Peptides (1997) 18(9):1423–9. doi:10.1016/S0196-9781(97)00231-3
7. Margolis RU, Altszuler N. Insulin in the cerebrospinal fluid. Nature (1967) 215(5108):1375–6. doi:10.1038/2151375a0
8. Woods SC, Porte D Jr. Relationship between plasma and cerebrospinal fluid insulin levels of dogs. Am J Physiol (1977) 233(4):E331–4.
9. Duffy KR, Pardridge WM. Blood-brain barrier transcytosis of insulin in developing rabbits. Brain Res (1987) 420(1):32–8. doi:10.1016/0006-8993(87)90236-8
10. Eckert GP, Kirsch C, Leutz S, Wood WG, Muller WE. Cholesterol modulates amyloid beta-peptide’s membrane interactions. Pharmacopsychiatry (2003) 36(Suppl 2):S136–43. doi:10.1055/s-2003-43059
11. Baura GD, Foster DM, Porte D Jr, Kahn SE, Bergman RN, Cobelli C, et al. Saturable transport of insulin from plasma into the central nervous system of dogs in vivo. A mechanism for regulated insulin delivery to the brain. J Clin Invest(1993) 92(4):1824–30. doi:10.1172/JCI116773
12. Banks WA, Kastin AJ. Differential permeability of the blood-brain barrier to two pancreatic peptides: insulin and amylin. Peptides (1998) 19(5):883–9. doi:10.1016/S0196-9781(98)00018-7
13. Baura GD, Foster DM, Kaiyala K, Porte D Jr, Kahn SE, Schwartz MW. Insulin transport from plasma into the central nervous system is inhibited by dexamethasone in dogs. Diabetes (1996) 45(1):86–90. doi:10.2337/diab.45.1.86
14. Strubbe JH, Porte D Jr, Woods SC. Insulin responses and glucose levels in plasma and cerebrospinal fluid during fasting and refeeding in the rat. Physiol Behav (1988) 44(2):205–8. doi:10.1016/0031-9384(88)90139-4
15. Kaiyala KJ, Prigeon RL, Kahn SE, Woods SC, Schwartz MW. Obesity induced by a high-fat diet is associated with reduced brain insulin transport in dogs. Diabetes (2000) 49(9):1525–33. doi:10.2337/diabetes.49.9.152
16. Florant GL, Richardson RD, Mahan S, Singer L, Woods SC. Seasonal changes in CSF insulin levels in marmots: insulin may not be a satiety signal for fasting in winter. Am J Physiol (1991) 260(4 Pt 2):R712–6.
17. Banks WA, Owen JB, Erickson MA. Insulin in the brain: there and back again. Pharmacol Ther (2012) 136(1):82–93. doi:10.1016/j.pharmthera.2012.07.006
18. Ghasemi R, Haeri A, Dargahi L, Mohamed Z, Ahmadiani A. Insulin in the brain: sources, localization and functions. Mol Neurobiol (2013) 47(1):145–71. doi:10.1007/s12035-012-8339-9
19. Dorn A, Rinne A, Bernstein HG, Hahn HJ, Ziegler M. Insulin and C-peptide in human brain neurons (insulin/C-peptide/brain peptides/immunohistochemistry/radioimmunoassay). J Hirnforsch (1983) 24(5):495–9.
20. Jezova D, Vigas M, Sadlon J. C-peptide-like material in rat brain: response to fasting and glucose ingestion. Endocrinol Exp (1985) 19(4):261–6.
21. Frolich L, Blum-Degen D, Bernstein HG, Engelsberger S, Humrich J, Laufer S, et al. Brain insulin and insulin receptors in aging and sporadic Alzheimer’s disease. J Neural Transm (1998) 105(4–5):423–38. doi:10.1007/s007020050068
22. Young WS III. Periventricular hypothalamic cells in the rat brain contain insulin mRNA. Neuropeptides (1986) 8(2):93–7. doi:10.1016/0143-4179(86)90035-1
23. Devaskar SU, Singh BS, Carnaghi LR, Rajakumar PA, Giddings SJ. Insulin II gene expression in rat central nervous system. Regul Pept (1993) 48(1–2):55–63. doi:10.1016/0167-0115(93)90335-6
24. Devaskar SU, Giddings SJ, Rajakumar PA, Carnaghi LR, Menon RK, Zahm DS. Insulin gene expression and insulin synthesis in mammalian neuronal cells. J Biol Chem (1994) 269(11):8445–54.
25. Raizada MK. Localization of insulin-like immunoreactivity in the neurons from primary cultures of rat brain. Exp Cell Res (1983) 143(2):351–7. doi:10.1016/0014-4827(83)90061-7
26. Birch NP, Christie DL, Renwick AG. Proinsulin-like material in mouse foetal brain cell cultures. FEBS Lett (1984) 168(2):299–302. doi:10.1016/0014-5793(84)80266-5
27. Schechter R, Whitmire J, Wheet GS, Beju D, Jackson KW, Harlow R, et al. Immunohistochemical and in situ hybridization study of an insulin-like substance in fetal neuron cell cultures. Brain Res (1994) 636(1):9–27. doi:10.1016/0006-8993(94)90170-8
28. Gerozissis K. Brain insulin: regulation, mechanisms of action and functions. Cell Mol Neurobiol (2003) 23(1):1–25. doi:10.1023/A:1025021529347
29. Clarke DW, Mudd L, Boyd FT Jr, Fields M, Raizada MK. Insulin is released from rat brain neuronal cells in culture. J Neurochem (1986) 47(3):831–6. doi:10.1111/j.1471-4159.1986.tb00686.x
30. Wei LT, Matsumoto H, Rhoads DE. Release of immunoreactive insulin from rat brain synaptosomes under depolarizing conditions. J Neurochem (1990) 54(5):1661–5. doi:10.1111/j.1471-4159.1990.tb01219.x
31. Santos MS, Pereira EM, Carvaho AP. Stimulation of immunoreactive insulin release by glucose in rat brain synaptosomes. Neurochem Res (1999) 24(1):33–6. doi:10.1023/A:1020971812098
32. Gerozissis K. Brain insulin, energy and glucose homeostasis; genes, environment and metabolic pathologies. Eur J Pharmacol (2008) 585(1):38–49. doi:10.1016/j.ejphar.2008.01.050
33. Burgos-Ramos E, Gonzalez-Rodriguez A, Canelles S, Baquedano E, Frago LM, Revuelta-Cervantes J, et al. Differential insulin receptor substrate-1 (IRS1)-related modulation of neuropeptide Y and proopiomelanocortin expression in nondiabetic and diabetic IRS2-/- mice. Endocrinology (2012) 153(3):1129–40. doi:10.1210/en.2011-1278
34. Miller DW, Keller BT, Borchardt RT. Identification and distribution of insulin receptors on cultured bovine brain microvessel endothelial cells: possible function in insulin processing in the blood-brain barrier. J Cell Physiol (1994) 161(2):333–41. doi:10.1002/jcp.1041610218
35. Tagliamonte A, DeMontis MG, Olianas M, Onali PL, Gessa GL. Possible role of insulin in the transport of tyrosine and tryptophan from blood to brain. Adv Exp Med Biol (1976) 69:89–94. doi:10.1007/978-1-4684-3264-0_7
36. Ayre SG, Skaletski B, Mosnaim AD. Blood-brain barrier passage of azidothymidine in rats: effect of insulin. Res Commun Chem Pathol Pharmacol (1989) 63(1):45–52.
37. Kastin AJ, Akerstrom V. Glucose and insulin increase the transport of leptin through the blood-brain barrier in normal mice but not in streptozotocin-diabetic mice. Neuroendocrinology (2001) 73(4):237–42. doi:10.1159/000054640
38. Liu H, Yang H, Wang D, Liu Y, Liu X, Li Y, et al. Insulin regulates P-glycoprotein in rat brain microvessel endothelial cells via an insulin receptor-mediated PKC/NF-kappaB pathway but not a PI3K/Akt pathway. Eur J Pharmacol (2009) 602(2–3):277–82. doi:10.1016/j.ejphar.2008.11.026
39. Liu X, Jing XY, Jin S, Li Y, Liu L, Yu YL, et al. Insulin suppresses the expression and function of breast cancer resistance protein in primary cultures of rat brain microvessel endothelial cells. Pharmacol Rep (2011) 63(2):487–93. doi:10.1016/S1734-1140(11)70515-1
40. Catalan RE, Martinez AM, Aragones MD, Miguel BG, Robles A. Insulin action on brain microvessels; effect on alkaline phosphatase. Biochem Biophys Res Commun (1988) 150(2):583–90. doi:10.1016/0006-291X(88)90433-0
41. Langston JW, Li W, Harrison L, Aw TY. Activation of promoter activity of the catalytic subunit of gamma-glutamylcysteine ligase (GCL) in brain endothelial cells by insulin requires antioxidant response element 4 and altered glycemic status: implication for GCL expression and GSH synthesis. Free Radic Biol Med (2011) 51(9):1749–57. doi:10.1016/j.freeradbiomed.2011.08.004
42. Hurley JH, Zhang S, Bye LS, Marshall MS, DePaoli-Roach AA, Guan K, et al. Insulin signaling inhibits the 5-HT2C receptor in choroid plexus via MAP kinase. BMC Neurosci (2003) 4:10. doi:10.1186/1471-2202-4-10
43. Bernstein HG, Lendeckel U, Bukowska A, Ansorge S, Ernst T, Stauch R, et al. Regional and cellular distribution patterns of insulin-degrading enzyme in the adult human brain and pituitary. J Chem Neuroanat (2008) 35(2):216–24. doi:10.1016/j.jchemneu.2007.12.001
44. Lynch JA, George AM, Eisenhauer PB, Conn K, Gao W, Carreras I, et al. Insulin degrading enzyme is localized predominantly at the cell surface of polarized and unpolarized human cerebrovascular endothelial cell cultures. J Neurosci Res (2006) 83(7):1262–70. doi:10.1002/jnr.20809
45. Seino S, Seino M, Nishi S, Bell GI. Structure of the human insulin receptor gene and characterization of its promoter. Proc Natl Acad Sci U S A (1989) 86(1):114–8. doi:10.1073/pnas.86.1.114
46. Moller DE, Yokota A, Caro JF, Flier JS. Tissue-specific expression of two alternatively spliced insulin receptor mRNAs in man. Mol Endocrinol (1989) 3(8):1263–9. doi:10.1210/mend-3-8-1263
47. Seino S, Bell GI. Alternative splicing of human insulin receptor messenger RNA. Biochem Biophys Res Commun (1989) 159(1):312–6. doi:10.1016/0006-291X(89)92439-X
48. Vienberg SG, Bouman SD, Sorensen H, Stidsen CE, Kjeldsen T, Glendorf T, et al. Receptor-isoform-selective insulin analogues give tissue-preferential effects. Biochem J (2011) 440(3):301–8. doi:10.1042/BJ20110880
49. Schulingkamp RJ, Pagano TC, Hung D, Raffa RB. Insulin receptors and insulin action in the brain: review and clinical implications. Neurosci Biobehav Rev (2000) 24(8):855–72. doi:10.1016/S0149-7634(00)00040-3
50. Szabo O, Szabo AJ. Evidence for an insulin-sensitive receptor in the central nervous system. Am J Physiol (1972) 223(6):1349–53.
51. Posner BI, Kelly PA, Shiu RP, Friesen HG. Studies of insulin, growth hormone and prolactin binding: tissue distribution, species variation and characterization. Endocrinology (1974) 95(2):521–31. doi:10.1210/endo-95-2-521
52. Havrankova J, Roth J, Brownstein M. Insulin receptors are widely distributed in the central nervous system of the rat. Nature (1978) 272(5656):827–9. doi:10.1038/272827a0
53. Lowe WL Jr, Boyd FT, Clarke DW, Raizada MK, Hart C, LeRoith D. Development of brain insulin receptors: structural and functional studies of insulin receptors from whole brain and primary cell cultures. Endocrinology (1986) 119(1):25–35. doi:10.1210/endo-119-1-25
54. Landau BR, Takaoka Y, Abrams MA, Genuth SM, van Houten M, Posner BI, et al. Binding of insulin by monkey and pig hypothalamus. Diabetes (1983) 32(3):284–91. doi:10.2337/diabetes.32.3.284
55. Hill JM, Lesniak MA, Pert CB, Roth J. Autoradiographic localization of insulin receptors in rat brain: prominence in olfactory and limbic areas. Neuroscience (1986) 17(4):1127–38. doi:10.1016/0306-4522(86)90082-5
56. Werther GA, Hogg A, Oldfield BJ, McKinley MJ, Figdor R, Allen AM, et al. Localization and characterization of insulin receptors in rat brain and pituitary gland using in vitro autoradiography and computerized densitometry. Endocrinology (1987) 121(4):1562–70. doi:10.1210/endo-121-4-1562
57. Marks JL, Porte D Jr, Stahl WL, Baskin DG. Localization of insulin receptor mRNA in rat brain by in situ hybridization. Endocrinology (1990) 127(6):3234–6. doi:10.1210/endo-127-6-3234
58. Amessou M, Tahiri K, Chauvet G, Desbuquois B. Age-related changes in insulin receptor mRNA and protein expression in genetically obese Zucker rats. Diabetes Metab (2010) 36(2):120–8. doi:10.1016/j.diabet.2009.09.004
59. Pacold ST, Blackard WG. Central nervous system insulin receptors in normal and diabetic rats. Endocrinology (1979) 105(6):1452–7.
60. Werther GA, Hogg A, Oldfield BJ, McKinley MJ, Figdor R, Mendelsohn FA. Localization and characterization of insulin-like growth factor-I receptors in rat brain and pituitary gland using in vitro autoradiography and computerized densitometry* A distinct distribution from insulin receptors. J Neuroendocrinol (1989) 1(5):369–77. doi:10.1111/j.1365-2826.1989.tb00131.x
61. Hami J, Sadr-Nabavi A, Sankian M, HaghИР H. Sex differences and left-right asymmetries in expression of insulin and insulin-like growth factor-1 receptors in developing rat hippocampus. Brain Struct Funct (2012) 217(2):293–302. doi:10.1007/s00429-011-0358-1
62. Hopkins DF, Williams G. Insulin receptors are widely distributed in human brain and bind human and porcine insulin with equal affinity. Diabet Med (1997) 14(12):1044–50. doi:10.1002/(SICI)1096-9136(199712)14:12<1044::AID-DIA508>3.3.CO;2-6
63. Potau N, Escofet MA, Martinez MC. Ontogenesis of insulin receptors in human cerebral cortex. J Endocrinol Invest(1991) 14(1):53–8. doi:10.1007/BF03350263
64. Zahniser NR, Goens MB, Hanaway PJ, Vinych JV. Characterization and regulation of insulin receptors in rat brain. J Neurochem (1984) 42(5):1354–62. doi:10.1111/j.1471-4159.1984.tb02795.x
65. Heidenreich KA, Zahniser NR, Berhanu P, Brandenburg D, Olefsky JM. Structural differences between insulin receptors in the brain and peripheral target tissues. J Biol Chem (1983) 258(14):8527–30.
66. Joost HG. Structural and functional heterogeneity of insulin receptors. Cell Signal (1995) 7(2):85–91. doi:10.1016/0898-6568(94)00071-I
67. Kar S, Chabot JG, Quirion R. Quantitative autoradiographic localization of [125I]insulin-like growth factor I, [125I]insulin-like growth factor II, and [125I]insulin receptor binding sites in developing and adult rat brain. J Comp Neurol (1993) 333(3):375–97. doi:10.1002/cne.903330306
68. Shemer J, Adamo M, Raizada MK, Heffez D, Zick Y, LeRoith D. Insulin and IGF-I stimulate phosphorylation of theИР respective receptors in intact neuronal and glial cells in primary culture. J Mol Neurosci (1989) 1(1):3–8. doi:10.1007/BF02918899
69. Brummer T, Schmitz-Peiffer C, Daly RJ. Docking proteins. FEBS J (2010) 277(21):4356–69. doi:10.1111/j.1742-4658.2010.07865.x
70. Taguchi A, White MF. Insulin-like signaling, nutrient homeostasis, and life span. Annu Rev Physiol (2008) 70:191–212. doi:10.1146/annurev.physiol.70.113006.100533
71. Saltiel AR, Pessin JE. Insulin signaling pathways in time and space. Trends Cell Biol (2002) 12(2):65–71. doi:10.1016/S0962-8924(01)02207-3
72. Pilch PF, Shia MA, Benson RJ, Fine RE. Coated vesicles participate in the receptor-mediated endocytosis of insulin. J Cell Biol (1983) 96(1):133–8. doi:10.1083/jcb.96.1.133
73. Heffetz D, Zick Y. Receptor aggregation is necessary for activation of the soluble insulin receptor kinase. J Biol Chem(1986) 261(2):889–94.
74. White MF. Insulin signaling in health and disease. Science (2003) 302(5651):1710–1. doi:10.1126/science.1092952
75. Myers MG Jr, Sun XJ, White MF. The IRS-1 signaling system. Trends Biochem Sci (1994) 19(7):289–93. doi:10.1016/0968-0004(94)90007-8
76. Aguirre V, Werner ED, Giraud J, Lee YH, Shoelson SE, White MF. Phosphorylation of Ser307 in insulin receptor substrate-1 blocks interactions with the insulin receptor and inhibits insulin action. J Biol Chem (2002) 277(2):1531–7. doi:10.1074/jbc.M101521200
77. Krebs DL, Hilton DJ. A new role for SOCS in insulin action. Suppressor of cytokine signaling. Sci STKE (2003) 2003(169):e6. doi:10.1126/stke.2003.169.pe6
78. Jhala US, Canettieri G, Screaton RA, Kulkarni RN, Krajewski S, Reed J, et al. cAMP promotes pancreatic beta-cell survival via CREB-mediated induction of IRS2. Genes Dev (2003) 17(13):1575–80. doi:10.1101/gad.1097103
79. Abbott MA, Wells DG, Fallon JR. The insulin receptor tyrosine kinase substrate p58/53 and the insulin receptor are components of CNS synapses. J Neurosci (1999) 19(17):7300–8.
80. Okamura-Oho Y, Miyashita T, Yamada M. Distinctive tissue distribution and phosphorylation of IRSp53 isoforms. Biochem Biophys Res Commun (2001) 289(5):957–60. doi:10.1006/bbrc.2001.6102
81. Miyahara A, Okamura-Oho Y, Miyashita T, Hoshika A, Yamada M. Genomic structure and alternative splicing of the insulin receptor tyrosine kinase substrate of 53-kDa protein. J Hum Genet (2003) 48(8):410–4. doi:10.1007/s10038-003-0047-x
82. Choi J, Ko J, Racz B, Burette A, Lee JR, Kim S, et al. Regulation of dendritic spine morphogenesis by insulin receptor substrate 53, a downstream effector of Rac1 and Cdc42 small GTPases. J Neurosci (2005) 25(4):869–79. doi:10.1523/JNEUROSCI.3212-04.2005
83. Mackie S, Aitken A. Novel brain 14-3-3 interacting proteins involved in neurodegenerative disease. FEBS J (2005) 272(16):4202–10. doi:10.1111/j.1742-4658.2005.04832.x
84. Sawallisch C, Berhorster K, Disanza A, Mantoani S, Kintscher M, Stoenica L, et al. The insulin receptor substrate of 53 kDa (IRSp53) limits hippocampal synaptic plasticity. J Biol Chem (2009) 284(14):9225–36. doi:10.1074/jbc.M808425200
85. Lizcano JM, Alessi DR. The insulin signalling pathway. Curr Biol (2002) 12(7):R236–8. doi:10.1016/S0960-9822(02)00777-7
86. Jewell JL, Guan KL. Nutrient signaling to mTOR and cell growth. Trends Biochem Sci (2013) 38(5):233–42. doi:10.1016/j.tibs.2013.01.004
87. Kolch W, Kotwaliwale A, Vass K, Janosch P. The role of Raf kinases in malignant transformation. Expert Rev Mol Med(2002) 4(8):1–18. doi:10.1017/S1462399402004386
88. Evans RM, Hui S, Perkins A, Lahiri DK, Poirier J, Farlow MR. Cholesterol and APOE genotype interact to influence Alzheimer disease progression. Neurology (2004) 62(10):1869–71. doi:10.1212/01.WNL.0000125323.15458.3F
89. Frodin M, Gammeltoft S. Role and regulation of 90 kDa ribosomal S6 kinase (RSK) in signal transduction. Mol Cell Endocrinol (1999) 151(1–2):65–77. doi:10.1016/S0303-7207(99)00061-1
90. White H, Venkatesh B. Clinical review: ketones and brain injury. Crit Care (2011) 15(2):219. doi:10.1186/cc10020
91. Marty N, Dallaporta M, Thorens B. Brain glucose sensing, counterregulation, and energy homeostasis. Physiology (Bethesda) (2007) 22:241–51. doi:10.1152/physiol.00010.2007
92. Alvarez E, Roncero I, Chowen JA, Thorens B, Blazquez E. Expression of the glucagon-like peptide-1 receptor gene in rat brain. J Neurochem (1996) 66(3):920–7. doi:10.1046/j.1471-4159.1996.66030920.x
93. Navarro M, Rodriquez de Fonseca F, Alvarez E, Chowen JA, Zueco JA, Gomez R, et al. Colocalization of glucagon-like peptide-1 (GLP-1) receptors, glucose transporter GLUT-2, and glucokinase mRNAs in rat hypothalamic cells: evidence for a role of GLP-1 receptor agonists as an inhibitory signal for food and water intake. J Neurochem (1996) 67(5):1982–91. doi:10.1046/j.1471-4159.1996.67051982.x
94. Roncero I, Alvarez E, Vazquez P, Blazquez E. Functional glucokinase isoforms are expressed in rat brain. J Neurochem(2000) 74(5):1848–57. doi:10.1046/j.1471-4159.2000.0741848.x
95. Thorens B, Mueckler M. Glucose transporters in the 21st century. Am J Physiol Endocrinol Metab (2010) 298(2):E141–5. doi:10.1152/ajpendo.00712.2009
96. Simpson IA, Appel NM, Hokari M, Oki J, Holman GD, Maher F, et al. Blood-brain barrier glucose transporter: effects of hypo- and hyper-glycemia revisited. J Neurochem (1999) 72(1):238–47. doi:10.1046/j.1471-4159.1999.0720238.x
97. Kang L, Routh VH, Kuzhikandathil EV, Gaspers LD, Levin BE. Physiological and molecular characteristics of rat hypothalamic ventromedial nucleus glucosensing neurons. Diabetes (2004) 53(3):549–59. doi:10.2337/diabetes.53.3.549
98. Levin BE, Routh VH, Kang L, Sanders NM, Dunn-Meynell AA. Neuronal glucosensing: what do we know after 50 years? Diabetes (2004) 53(10):2521–8. doi:10.2337/diabetes.53.10.2521
99. Li B, Xi X, Roane DS, Ryan DH, Martin RJ. Distribution of glucokinase, glucose transporter GLUT2, sulfonylurea receptor-1, glucagon-like peptide-1 receptor and neuropeptide Y messenger RNAs in rat brain by quantitative real time RT-PCR. Brain Res Mol Brain Res (2003) 113(1–2):139–42. doi:10.1016/S0169-328X(03)00125-6
100. Nagamatsu S, Kornhauser JM, Burant CF, Seino S, Mayo KE, Bell GI. Glucose transporter expression in brain. cDNA sequence of mouse GLUT3, the brain facilitative glucose transporter isoform, and identification of sites of expression by in situ hybridization. J Biol Chem (1992) 267(1):467–72.
101. Gould GW, Brant AM, Kahn BB, Shepherd PR, McCoid SC, Gibbs EM. Expression of the brain-type glucose transporter is restricted to brain and neuronal cells in mice. Diabetologia (1992) 35(4):304–9. doi:10.1007/BF00401196
102. El Messari S, Leloup C, Quignon M, Brisorgueil MJ, Penicaud L, Arluison M. Immunocytochemical localization of the insulin-responsive glucose transporter 4 (Glut4) in the rat central nervous system. J Comp Neurol (1998) 399(4):492–512. doi:10.1002/(SICI)1096-9861(19981005)399:4<492::AID-CNE4>3.0.CO;2-X
103. Vannucci SJ, Koehler-Stec EM, Li K, Reynolds TH, Clark R, Simpson IA. GLUT4 glucose transporter expression in rodent brain: effect of diabetes. Brain Res (1998) 797(1):1–11. doi:10.1016/S0006-8993(98)00103-6
104. Talbot K, Wang HY, Kazi H, Han LY, Bakshi KP, Stucky A, et al. Demonstrated brain insulin resistance in Alzheimer’s disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. J Clin Invest (2012) 122(4):1316–38. doi:10.1172/JCI59903
105. Livingstone C, Lyall H, Gould GW. Hypothalamic GLUT 4 expression: a glucose- and insulin-sensing mechanism? Mol Cell Endocrinol (1995) 107(1):67–70. doi:10.1016/0303-7207(94)03423-Q
106. Wang R, Liu X, Hentges ST, Dunn-Meynell AA, Levin BE, Wang W, et al. The regulation of glucose-excited neurons in the hypothalamic arcuate nucleus by glucose and feeding-relevant peptides. Diabetes (2004) 53(8):1959–65. doi:10.2337/diabetes.53.8.1959
107. Reagan LP, Gorovits N, Hoskin EK, Alves SE, Katz EB, Grillo CA, et al. Localization and regulation of GLUTx1 glucose transporter in the hippocampus of streptozotocin diabetic rats. Proc Natl Acad Sci U S A (2001) 98(5):2820–5. doi:10.1073/pnas.051629798
108. Ibberson M, Riederer BM, Uldry M, Guhl B, Roth J, Thorens B. Immunolocalization of GLUTX1 in the testis and to specific brain areas and vasopressin-containing neurons. Endocrinology (2002) 143(1):276–84. doi:10.1210/endo.143.1.8587
109. Reagan LP, Rosell DR, Alves SE, Hoskin EK, McCall AL, Charron MJ, et al. GLUT8 glucose transporter is localized to excitatory and inhibitory neurons in the rat hippocampus. Brain Res (2002) 932(1–2):129–34. doi:10.1016/S0006-8993(02)02308-9
110. Sankar R, Thamotharan S, Shin D, Moley KH, Devaskar SU. Insulin-responsive glucose transporters-GLUT8 and GLUT4 are expressed in the developing mammalian brain. Brain Res Mol Brain Res (2002) 107(2):157–65. doi:10.1016/S0169-328X(02)00487-4
111. McEwen BS, Reagan LP. Glucose transporter expression in the central nervous system: relationship to synaptic function. Eur J Pharmacol (2004) 490(1–3):13–24. doi:10.1016/j.ejphar.2004.02.041
112. Piroli GG, Grillo CA, Hoskin EK, Znamensky V, Katz EB, Milner TA, et al. Peripheral glucose administration stimulates the translocation of GLUT8 glucose transporter to the endoplasmic reticulum in the rat hippocampus. J Comp Neurol(2002) 452(2):103–14. doi:10.1002/cne.10368
113. Obici S, Feng Z, Karkanias G, Baskin DG, Rossetti L. Decreasing hypothalamic insulin receptors causes hyperphagia and insulin resistance in rats. Nat Neurosci (2002) 5(6):566–72. doi:10.1038/nn0602-861
114. Obici S, Zhang BB, Karkanias G, Rossetti L. Hypothalamic insulin signaling is required for inhibition of glucose production. Nat Med (2002) 8(12):1376–82. doi:10.1038/nm1202-798
115. Demuro G, Obici S. Central nervous system and control of endogenous glucose production. Curr Diab Rep (2006) 6(3):188–93. doi:10.1007/s11892-006-0033-8
116. Girard J. The inhibitory effects of insulin on hepatic glucose production are both direct and indirect. Diabetes (2006) 55(Suppl 2):S65–9. doi:10.2337/db06-S009
117. Pocai A, Lam TK, Gutierrez-Juarez R, Obici S, Schwartz GJ, Bryan J, et al. Hypothalamic K(ATP) channels control hepatic glucose production. Nature (2005) 434(7036):1026–31. doi:10.1038/nature03439
118. Cotero VE, Routh VH. Insulin blunts the response of glucose-excited neurons in the ventrolateral-ventromedial hypothalamic nucleus to decreased glucose. Am J Physiol Endocrinol Metab (2009) 296(5):E1101–9. doi:10.1152/ajpendo.90932.2008
119. Bernard C. Leçons de Physiologie Expérimentale Appliquée à la Médecine. Paris: Baillére (1855).
120. Niswender KD, Morrison CD, Clegg DJ, Olson R, Baskin DG, Myers MG Jr, et al. Insulin activation of phosphatidylinositol 3-kinase in the hypothalamic arcuate nucleus: a key mediator of insulin-induced anorexia. Diabetes (2003) 52(2):227–31. doi:10.2337/diabetes.52.2.227
121. Kim YB, Uotani S, Pierroz DD, Flier JS, Kahn BB. In vivo administration of leptin activates signal transduction directly in insulin-sensitive tissues: overlapping but distinct pathways from insulin. Endocrinology (2000) 141(7):2328–39. doi:10.1210/endo.141.7.7536
122. Castellano JM, Roa J, Luque RM, Dieguez C, Aguilar E, Pinilla L, et al. KiSS-1/kisspeptins and the metabolic control of reproduction: physiologic roles and putative physiopathological implications. Peptides (2009) 30(1):139–45. doi:10.1016/j.peptides.2008.06.007
123. Fernandez-Fernandez R, Martini AC, Navarro VM, Castellano JM, Dieguez C, Aguilar E, et al. Novel signals for the integration of energy balance and reproduction. Mol Cell Endocrinol (2006) 25(4–255):127–32. doi:10.1016/j.mce.2006.04.026
124. Arias P, Rodriguez M, Szwarcfarb B, Sinay IR, Moguilevsky JA. Effect of insulin on LHRH release by perifused hypothalamic fragments. Neuroendocrinology (1992) 56(3):415–8. doi:10.1159/000126257
125. Miller DW, Blache D, Martin GB. The role of intracerebral insulin in the effect of nutrition on gonadotrophin secretion in mature male sheep. J Endocrinol (1995) 147(2):321–9. doi:10.1677/joe.0.1470321
126. Dong Q, Lazarus RM, Wong LS, Vellios M, Handelsman DJ. Pulsatile LH secretion in streptozotocin-induced diabetes in the rat. J Endocrinol (1991) 131(1):49–55. doi:10.1677/joe.0.1310049
127. Tanaka T, Nagatani S, Bucholtz DC, Ohkura S, Tsukamura H, Maeda K, et al. Central action of insulin regulates pulsatile luteinizing hormone secretion in the diabetic sheep model. Biol Reprod (2000) 62(5):1256–61. doi:10.1095/biolreprod62.5.1256
128. Bucholtz DC, Chiesa A, Pappano WN, Nagatani S, Tsukamura H, Maeda KI, et al. Regulation of pulsatile luteinizing hormone secretion by insulin in the diabetic male lamb. Biol Reprod (2000) 62(5):1248–55. doi:10.1095/biolreprod62.5.1248
129. Roger LJ, Fellows RE. Stimulation of ornithine decarboxylase activity by insulin in developing rat brain. Endocrinology(1980) 106(2):619–25. doi:10.1210/endo-106-2-619
130. Wozniak M, Rydzewski B, Baker SP, Raizada MK. The cellular and physiological actions of insulin in the central nervous system. Neurochem Int (1993) 22(1):1–10. doi:10.1016/0197-0186(93)90062-A
131. Schubert M, Brazil DP, Burks DJ, Kushner JA, Ye J, Flint CL, et al. Insulin receptor substrate-2 deficiency impairs brain growth and promotes tau phosphorylation. J Neurosci (2003) 23(18):7084–92.
132. Xu QG, Li XQ, Kotecha SA, Cheng C, Sun HS, Zochodne DW. Insulin as an in vivo growth factor. Exp Neurol (2004) 188(1):43–51. doi:10.1016/j.expneurol.2004.03.008
133. Raizada MK, Yang JW, Fellows RE. Binding of [125I]insulin to specific receptors and stimulation of nucleotide incorporation in cells cultured from rat brain. Brain Res (1980) 200(2):389–400. doi:10.1016/0006-8993(80)90929-4
134. Heidenreich KA, de Vellis G, Gilmore PR. Functional properties of the subtype of insulin receptor found on neurons. J Neurochem (1988) 51(3):878–87. doi:10.1111/j.1471-4159.1988.tb01824.x
135. Clarke DW, Boyd FT Jr, Kappy MS, Raizada MK. Insulin stimulates macromolecular synthesis in cultured glial cells from rat brain. Am J Physiol (1985) 249(5 Pt 1):C484–9.
136. Heidenreich KA, Toledo SP. Insulin receptors mediate growth effects in cultured fetal neurons. II. Activation of a protein kinase that phosphorylates ribosomal protein S6. Endocrinology (1989) 125(3):1458–63. doi:10.1210/endo-125-3-1451
137. Recio-Pinto E, Lang FF, Ishii DN. Insulin and insulin-like growth factor II permit nerve growth factor binding and the neurite formation response in cultured human neuroblastoma cells. Proc Natl Acad Sci U S A (1984) 81(8):2562–6. doi:10.1073/pnas.81.8.2562
138. Ang LC, Bhaumick B, Juurlink BH. Neurite promoting activity of insulin, insulin-like growth factor I and nerve growth factor on spinal motoneurons is astrocyte dependent. Brain Res Dev Brain Res (1993) 74(1):83–8. doi:10.1016/0165-3806(93)90086-P
139. Velazquez E, Blazquez E, Ruiz-Albusac JM. Synergistic effect of glucagon-like peptide 2 (GLP-2) and of key growth factors on the proliferation of cultured rat astrocytes. Evidence for reciprocal upregulation of the mRNAs for GLP-2 and IGF-I receptors. Mol Neurobiol (2009) 40(2):183–93. doi:10.1007/s12035-009-8080-1
140. Heni M, Hennige AM, Peter A, Siegel-Axel D, Ordelheide AM, Krebs N, et al. Insulin promotes glycogen storage and cell proliferation in primary human astrocytes. PLoS One (2011) 6(6):e21594. doi:10.1371/journal.pone.0021594
141. Heidenreich KA, Toledo SP, Brunton LL, Watson MJ, Daniel-Issakani S, Strulovici B. Insulin stimulates the activity of a novel protein kinase C, PKC-epsilon, in cultured fetal chick neurons. J Biol Chem (1990) 265(25):15076–82.
142. Vanhems E, Delbos M, Girardie J. Insulin and neuroparsin promote neurite outgrowth in cultured locust CNS. Eur J Neurosci (1990) 2(9):776–82. doi:10.1111/j.1460-9568.1990.tb00468.x
143. Heidenreich KA, Toledo SP, Kenner KA. Regulation of protein phosphorylation by insulin and insulin-like growth factors in cultured fetal neurons. Adv Exp Med Biol (1991) 293:379–84. doi:10.1007/978-1-4684-5949-4_33
144. Patel RA, Kurian P, Raizada MK, Crews FT. Insulin stimulates phosphatidylinositol 3-kinase activity in rat neuronal primary cultures. J Neurochem (1993) 61(1):360–3. doi:10.1111/j.1471-4159.1993.tb03578.x
145. Lee CC, Huang CC, Wu MY, Hsu KS. Insulin stimulates postsynaptic density-95 protein translation via the phosphoinositide 3-kinase-Akt-mammalian target of rapamycin signaling pathway. J Biol Chem (2005) 280(18):18543–50. doi:10.1074/jbc.M414112200
146. Lee CC, Huang CC, Hsu KS. Insulin promotes dendritic spine and synapse formation by the PI3K/Akt/mTOR and Rac1 signaling pathways. Neuropharmacology (2011) 61(4):867–79. doi:10.1016/j.neuropharm.2011.06.003
147. Nemoto T, Yanagita T, Satoh S, Maruta T, Kanai T, Murakami M, et al. Insulin-induced neurite-like process outgrowth: acceleration of tau protein synthesis via a phosphoinositide 3-kinase mammalian target of rapamycin pathway. Neurochem Int (2011) 59(6):880–8. doi:10.1016/j.neuint.2011.08.002
148. Schechter R, Yanovitch T, Abboud M, Johnson G III, Gaskins J. Effects of brain endogenous insulin on neurofilament and MAPK in fetal rat neuron cell cultures. Brain Res (1998) 808(2):270–8. doi:10.1016/S0006-8993(98)00842-7
149. Rhee YH, Choi M, Lee HS, Park CH, Kim SM, Yi SH, et al. Insulin concentration is critical in culturing human neural stem cells and neurons. Cell Death Dis (2013) 4:e766. doi:10.1038/cddis.2013.295
150. Leopold P. Neuronal differentiation: TOR and insulin receptor pathways set the tempo. Cell (2004) 119(1):4–5. doi:10.1016/j.cell.2004.09.024
151. Ryu BR, Ko HW, Jou I, Noh JS, Gwag BJ. Phosphatidylinositol 3-kinase-mediated regulation of neuronal apoptosis and necrosis by insulin and IGF-I. J Neurobiol (1999) 39(4):536–46. doi:10.1002/(SICI)1097-4695(19990615)39:4<536::AID-NEU7>3.3.CO;2-A
152. Rensink AA, Otte-Holler I, de Boer R, Bosch RR, ten Donkelaar HJ, de Waal RM, et al. Insulin inhibits amyloid beta-induced cell death in cultured human brain pericytes. Neurobiol Aging (2004) 25(1):93–103. doi:10.1016/S0197-4580(03)00039-3
153. Garg R, Chaudhuri A, Munschauer F, Dandona P. Hyperglycemia, insulin, and acute ischemic stroke: a mechanistic justification for a trial of insulin infusion therapy. Stroke (2006) 37(1):267–73. doi:10.1161/01.STR.0000195175.29487.30
154. Duarte AI, Proenca T, Oliveira CR, Santos MS, Rego AC. Insulin restores metabolic function in cultured cortical neurons subjected to oxidative stress. Diabetes (2006) 55(10):2863–70. doi:10.2337/db06-0030
155. Duarte AI, Santos MS, Seica R, de Oliveira CR. Insulin affects synaptosomal GABA and glutamate transport under oxidative stress conditions. Brain Res (2003) 977(1):23–30. doi:10.1016/S0006-8993(03)02679-9
156. Sevanian A, Davies KJ, Hochstein P. Serum urate as an antioxidant for ascorbic acid. Am J Clin Nutr (1991) 54(6 Suppl):1129S–34S.
157. Auer RN. Insulin, blood glucose levels, and ischemic brain damage. Neurology (1998) 51(3 Suppl 3):S39–43. doi:10.1212/WNL.51.3_Suppl_3.S39
158. Shuaib A, Ijaz MS, Waqar T, Voll C, Kanthan R, Miyashita H, et al. Insulin elevates hippocampal GABA levels during ischemia. This is independent of its hypoglycemic effect. Neuroscience (1995) 67(4):809–14. doi:10.1016/0306-4522(95)00093-X
159. Grunstein HS, James DE, Storlien LH, Smythe GA, Kraegen EW. Hyperinsulinemia suppresses glucose utilization in specific brain regions: in vivo studies using the euglycemic clamp in the rat. Endocrinology (1985) 116(2):604–10. doi:10.1210/endo-116-2-604
160. Voll CL, Auer RN. Insulin attenuates ischemic brain damage independent of its hypoglycemic effect. J Cereb Blood Flow Metab (1991) 11(6):1006–14. doi:10.1038/jcbfm.1991.168
161. Kovacs P, Hajnal A. In vivo electrophysiological effects of insulin in the rat brain. Neuropeptides (2009) 43(4):283–93. doi:10.1016/j.npep.2009.05.006
162. Wang Q, Liu L, Pei L, Ju W, Ahmadian G, Lu J, et al. Control of synaptic strength, a novel function of Akt. Neuron(2003) 38(6):915–28. doi:10.1016/S0896-6273(03)00356-8
163. Plum L, Schubert M, Bruning JC. The role of insulin receptor signaling in the brain. Trends Endocrinol Metab (2005) 16(2):59–65. doi:10.1016/j.tem.2005.01.008
164. Jonas EA, Knox RJ, Smith TC, Wayne NL, Connor JA, Kaczmarek LK. Regulation by insulin of a unique neuronal Ca2+ pool and of neuropeptide secretion. Nature (1997) 385(6614):343–6. doi:10.1038/385343a0
165. Boyd FT Jr, Clarke DW, Muther TF, Raizada MK. Insulin receptors and insulin modulation of norepinephrine uptake in neuronal cultures from rat brain. J Biol Chem (1985) 260(29):15880–4.
166. Masters BA, Shemer J, Judkins JH, Clarke DW, Le Roith D, Raizada MK. Insulin receptors and insulin action in dissociated brain cells. Brain Res (1987) 417(2):247–56. doi:10.1016/0006-8993(87)90449-5
167. Lozovsky D, Saller CF, Kopin IJ. Dopamine receptor binding is increased in diabetic rats. Science (1981) 214(4524):1031–3. doi:10.1126/science.6458088
168. Lozovsky DB, Kopin IJ, Saller CF. Modulation of dopamine receptor supersensitivity by chronic insulin: implication in schizophrenia. Brain Res (1985) 343(1):190–3. doi:10.1016/0006-8993(85)91178-3
169. Levin BE, Israel P, Lattemann DP. Insulin selectively downregulates alpha2-adrenoceptors in the arcuate and dorsomedial nucleus. Brain Res Bull (1998) 45(2):179–81. doi:10.1016/S0361-9230(97)00336-5
170. Rhoads DE, DiRocco RJ, Osburn LD, Peterson NA, Raghupathy E. Stimulation of synaptosomal uptake of neurotransmitter amino acids by insulin: possible role of insulin as a neuromodulator. Biochem Biophys Res Commun(1984) 119(3):1198–204. doi:10.1016/0006-291X(84)90903-3
171. Park CR, Seeley RJ, Craft S, Woods SC. Intracerebroventricular insulin enhances memory in a passive-avoidance task. Physiol Behav (2000) 68(4):509–14. doi:10.1016/S0031-9384(99)00220-6
172. Zhao W, Chen H, Xu H, Moore E, Meiri N, Quon MJ, et al. Brain insulin receptors and spatial memory. Correlated changes in gene expression, tyrosine phosphorylation, and signaling molecules in the hippocampus of water maze trained rats. J Biol Chem (1999) 274(49):34893–902. doi:10.1074/jbc.274.49.34893
173. Voll CL, Whishaw IQ, Auer RN. Postischemic insulin reduces spatial learning deficit following transient forebrain ischemia in rats. Stroke (1989) 20(5):646–51. doi:10.1161/01.STR.20.5.646
174. Lannert H, Hoyer S. Intracerebroventricular administration of streptozotocin causes long-term diminutions in learning and memory abilities and in cerebral energy metabolism in adult rats. Behav Neurosci (1998) 112(5):1199–208. doi:10.1037/0735-7044.112.5.1199
175. Kern W, Peters A, Fruehwald-Schultes B, Deininger E, Born J, Fehm HL. Improving influence of insulin on cognitive functions in humans. Neuroendocrinology (2001) 74(4):270–80. doi:10.1159/000054694
176. Feldman DE. Synaptic mechanisms for plasticity in neocortex. Annu Rev Neurosci (2009) 32:33–55. doi:10.1146/annurev.neuro.051508.135516
177. Song I, HuganИР RL. Regulation of AMPA receptors during synaptic plasticity. Trends Neurosci (2002) 25(11):578–88. doi:10.1016/S0166-2236(02)02270-1
178. Huang CC, Lee CC, Hsu KS. An investigation into signal transduction mechanisms involved in insulin-induced long-term depression in the CA1 region of the hippocampus. J Neurochem (2004) 89(1):217–31. doi:10.1111/j.1471-4159.2003.02307.x
179. Ahmadian G, Ju W, Liu L, Wyszynski M, Lee SH, Dunah AW, et al. Tyrosine phosphorylation of GluR2 is required for insulin-stimulated AMPA receptor endocytosis and LTD. EMBO J (2004) 23(5):1040–50. doi:10.1038/sj.emboj.7600126
180. Choopani S, Moosavi M, Naghdi N. Involvement of nitric oxide in insulin induced memory improvement. Peptides(2008) 29(6):898–903. doi:10.1016/j.peptides.2008.01.005
181. Ramsey MM, Adams MM, Ariwodola OJ, Sonntag WE, Weiner JL. Functional characterization of des-IGF-1 action at excitatory synapses in the CA1 region of rat hippocampus. J Neurophysiol (2005) 94(1):247–54. doi:10.1152/jn.00768.2004
182. Le Grevès M, Zhou Q, Berg M, Le Grevès P, Fhölenhag K, Meyerson B, et al. Growth hormone replacement in hypophysectomized rats affects spatial performance and hippocampal levels of NMDA receptor subunit and PSD-95 gene transcript levels. Exp Brain Res (2006) 173(2):267–73. doi:10.1007/s00221-006-0438-2
183. Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, et al. Inflammation and Alzheimer’s disease. Neurobiol Aging (2000) 21(3):383–421. doi:10.1016/S0197-4580(00)00124-X
184. Montine TJ, Kaye JA, Montine KS, McFarland L, Morrow JD, Quinn JF. Cerebrospinal fluid abeta42, tau, and f2-isoprostane concentrations in patients with Alzheimer disease, other dementias, and in age-matched controls. Arch Pathol Lab Med (2001) 125(4):510–2. doi:10.1043/0003-9985(2001)125<0510:CFATAF>2.0.CO;2
185. Sheng JG, Bora SH, Xu G, Borchelt DR, Price DL, Koliatsos VE. Lipopolysaccharide-induced-neuroinflammation increases intracellular accumulation of amyloid precursor protein and amyloid beta peptide in APPswe transgenic mice. Neurobiol Dis (2003) 14(1):133–45. doi:10.1016/S0969-9961(03)00069-X
186. Dandona P. Endothelium, inflammation, and diabetes. Curr Diab Rep (2002) 2(4):311–5. doi:10.1007/s11892-002-0019-0
187. Soop M, Duxbury H, Agwunobi AO, Gibson JM, Hopkins SJ, Childs C, et al. Euglycemic hyperinsulinemia augments the cytokine and endocrine responses to endotoxin in humans. Am J Physiol Endocrinol Metab (2002) 282(6):E1276–85. doi:10.1152/ajpendo.00535.2001
188. Fishel MA, Watson GS, Montine TJ, Wang Q, Green PS, Kulstad JJ, et al. Hyperinsulinemia provokes synchronous increases in central inflammation and beta-amyloid in normal adults. Arch Neurol (2005) 62(10):1539–44. doi:10.1001/archneur.62.10.noc50112
189. Johnston H, Boutin H, Allan SM. Assessing the contribution of inflammation in models of Alzheimer’s disease. Biochem Soc Trans (2011) 39(4):886–90. doi:10.1042/BST0390886
190. Rogers J, Mastroeni D, Leonard B, Joyce J, Grover A. Neuroinflammation in Alzheimer’s disease and Parkinson’s disease: are microglia pathogenic in either disorder? Int Rev Neurobiol (2007) 82:235–46. doi:10.1016/S0074-7742(07)82012-5
191. Pistell PJ, Morrison CD, Gupta S, Knight AG, Keller JN, Ingram DK, et al. Cognitive impairment following high fat diet consumption is associated with brain inflammation. J Neuroimmunol (2010) 219(1–2):25–32. doi:10.1016/j.jneuroim.2009.11.010
192. Neumann KF, Rojo L, Navarrete LP, Farias G, Reyes P, Maccioni RB. Insulin resistance and Alzheimer’s disease: molecular links & clinical implications. Curr Alzheimer Res (2008) 5(5):438–47. doi:10.2174/156720508785908919
193. Zhao M, Cribbs DH, Anderson AJ, Cummings BJ, Su JH, Wasserman AJ, et al. The induction of the TNFalpha death domain signaling pathway in Alzheimer’s disease brain. Neurochem Res (2003) 28(2):307–18. doi:10.1023/A:1022337519035
194. Dzienis-Straczkowska S, Straczkowski M, Szelachowska M, Stepien A, Kowalska I, Kinalska I. Soluble tumor necrosis factor-alpha receptors in young obese subjects with normal and impaired glucose tolerance. Diabetes Care (2003) 26(3):875–80. doi:10.2337/diacare.26.3.875
195. Bastard JP, Jardel C, Bruckert E, Vidal H, Hainque B. Variations in plasma soluble tumour necrosis factor receptors after diet-induced weight loss in obesity. Diabetes Obes Metab (2000) 2(5):323–5. doi:10.1046/j.1463-1326.2000.00090.x
196. Wrighten SA, Piroli GG, Grillo CA, Reagan LP. A look inside the diabetic brain: contributors to diabetes-induced brain aging. Biochim Biophys Acta (2009) 1792(5):444–53. doi:10.1016/j.bbadis.2008.10.013
197. Ghasemi R, Dargahi L, Haeri A, Moosavi M, Mohamed Z, Ahmadiani A. Brain insulin dysregulation: implication for neurological and neuropsychiatric disorders. Mol Neurobiol (2013) 47(3):1045–65. doi:10.1007/s12035-013-8404-z
198. Avila J, Wandosell F, Hernandez F. Role of glycogen synthase kinase-3 in Alzheimer’s disease pathogenesis and glycogen synthase kinase-3 inhibitors. Expert Rev Neurother (2010) 10(5):703–10. doi:10.1586/ern.10.40
199. Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature (1995) 378(6559):785–9. doi:10.1038/378785a0
200. Lee J, Kim MS. The role of GSK3 in glucose homeostasis and the development of insulin resistance. Diabetes Res Clin Pract (2007) 77(Suppl 1):S49–57. doi:10.1016/j.diabres.2007.01.033
201. Beurel E, Jope RS. Differential regulation of STAT family members by glycogen synthase kinase-3. J Biol Chem (2008) 283(32):21934–44. doi:10.1074/jbc.M802481200
202. Martin M, Rehani K, Jope RS, Michalek SM. Toll-like receptor-mediated cytokine production is differentially regulated by glycogen synthase kinase 3. Nat Immunol (2005) 6(8):777–84. doi:10.1038/ni1221
203. Agostinho P, Cunha RA, Oliveira C. Neuroinflammation, oxidative stress and the pathogenesis of Alzheimer’s disease. Curr Pharm Des (2010) 16(25):2766–78. doi:10.2174/138161210793176572
204. Najem D, Bamji-Mirza M, Chang N, Liu QY, Zhang W. Insulin resistance, neuroinflammation, and Alzheimer’s disease. Rev Neurosci (2014) 25(4):509–25. doi:10.1515/revneuro-2013-0050
205. Olefsky JM, Glass CK. Macrophages, inflammation, and insulin resistance. Annu Rev Physiol (2010) 72:219–46. doi:10.1146/annurev-physiol-021909-135846
206. Lee YS, Li P, Huh JY, Hwang IJ, Lu M, Kim JI, et al. Inflammation is necessary for long-term but not short-term high-fat diet-induced insulin resistance. Diabetes (2011) 60(10):2474–83. doi:10.2337/db11-0194
207. Hotamisligil GS, Budavari A, Murray D, Spiegelman BM. Reduced tyrosine kinase activity of the insulin receptor in obesity-diabetes. Central role of tumor necrosis factor-alpha. J Clin Invest (1994) 94(4):1543–9.
208. Kalupahana NS, Moustaid-Moussa N. The renin-angiotensin system: a link between obesity, inflammation and insulin resistance. Obes Rev (2012) 13(2):136–49. doi:10.1111/j.1467-789X.2011.00942.x
209. Wellen KE, Hotamisligil GS. Inflammation, stress, and diabetes. J Clin Invest (2005) 115(5):1111–9. doi:10.1172/JCI25102
210. Tu YF, Tsai YS, Wang LW, Wu HC, Huang CC, Ho CJ. Overweight worsens apoptosis, neuroinflammation and blood-brain barrier damage after hypoxic ischemia in neonatal brain through JNK hyperactivation. J Neuroinflammation(2011) 8:40. doi:10.1186/1742-2094-8-40
211. Sartorius T, Lutz SZ, Hoene M, Waak J, Peter A, Weigert C, et al. Toll-like receptors 2 and 4 impaИР insulin-mediated brain activity by interleukin-6 and osteopontin and alter sleep architecture. FASEB J (2012) 26(5):1799–809. doi:10.1096/fj.11-191023
212. Arruda AP, Milanski M, Coope A, Torsoni AS, Ropelle E, Carvalho DP, et al. Low-grade hypothalamic inflammation leads to defective thermogenesis, insulin resistance, and impaired insulin secretion. Endocrinology (2011) 152(4):1314–26. doi:10.1210/en.2010-0659
213. Konner AC, Bruning JC. Toll-like receptors: linking inflammation to metabolism. Trends Endocrinol Metab (2011) 22(1):16–23. doi:10.1016/j.tem.2010.08.007
214. Frolich L, Blum-Degen D, Riederer P, Hoyer S. A disturbance in the neuronal insulin receptor signal transduction in sporadic Alzheimer’s disease. Ann N Y Acad Sci (1999) 893:290–3. doi:10.1111/j.1749-6632.1999.tb07839.x
215. Zhao WQ, Lacor PN, Chen H, Lambert MP, Quon MJ, Krafft GA, et al. Insulin receptor dysfunction impairs cellular clearance of neurotoxic oligomeric a{beta}. J Biol Chem (2009) 284(28):18742–53. doi:10.1074/jbc.M109.011015
216. Xie L, Helmerhorst E, Taddei K, Plewright B, Van Bronswijk W, Martins R. Alzheimer’s beta-amyloid peptides compete for insulin binding to the insulin receptor. J Neurosci (2002) 22(10):RC221.
217. Lee HK, Kumar P, Fu Q, Rosen KM, Querfurth HW. The insulin/Akt signaling pathway is targeted by intracellular beta-amyloid. Mol Biol Cell (2009) 20(5):1533–44. doi:10.1091/mbc.E08-07-0777
218. Holscher C. Diabetes as a risk factor for Alzheimer’s disease: insulin signalling impairment in the brain as an alternative model of Alzheimer’s disease. Biochem Soc Trans (2011) 39(4):891–7. doi:10.1042/BST0390891
219. Bosco D, Fava A, Plastino M, Montalcini T, Pujia A. Possible implications of insulin resistance and glucose metabolism in Alzheimer’s disease pathogenesis. J Cell Mol Med (2011) 15(9):1807–21. doi:10.1111/j.1582-4934.2011.01318.x
220. Citron M. Alzheimer’s disease: treatments in discovery and development. Nat Neurosci (2002) 5(Suppl):1055–7. doi:10.1038/nn940
221. Correia SC, Santos RX, Carvalho C, Cardoso S, Candeias E, Santos MS, et al. Insulin signaling, glucose metabolism and mitochondria: major players in Alzheimer’s disease and diabetes interrelation. Brain Res (2012) 1441:64–78. doi:10.1016/j.brainres.2011.12.063
222. Efendic S, Luft R, Wajngot A. Aspects of the pathogenesis of type 2 diabetes. Endocr Rev (1984) 5(3):395–410. doi:10.1210/edrv-5-3-395
223. Chen Z, Zhong C. Decoding Alzheimer’s disease from perturbed cerebral glucose metabolism: implications for diagnostic and therapeutic strategies. Prog Neurobiol (2013) 108:21–43. doi:10.1016/j.pneurobio.2013.06.004
224. Janson J, Ashley RH, Harrison D, McIntyre S, Butler PC. The mechanism of islet amyloid polypeptide toxicity is membrane disruption by intermediate-sized toxic amyloid particles. Diabetes (1999) 48(3):491–8. doi:10.2337/diabetes.48.3.491
225. Luchsinger JA, Tang MX, Shea S, Mayeux R. Hyperinsulinemia and risk of Alzheimer disease. Neurology (2004) 63(7):1187–92. doi:10.1212/01.WNL.0000140292.04932.87
226. Janson J, Laedtke T, Parisi JE, O’Brien P, Petersen RC, Butler PC. Increased risk of type 2 diabetes in Alzheimer disease. Diabetes (2004) 53(2):474–81. doi:10.2337/diabetes.53.2.474
227. Hokama M, Oka S, Leon J, Ninomiya T, Honda H, Sasaki K, et al. Altered expression of diabetes-related genes in Alzheimer’s disease brains: the Hisayama study. Cereb Cortex (2013) 24(9):2476–88. doi:10.1093/cercor/bht101
228. de la Monte SM, Wands JR. Alzheimer’s disease is type 3 diabetes-evidence reviewed. J Diabetes Sci Technol (2008) 2(6):1101–13. doi:10.1177/193229680800200619
229. Correia SC, Santos RX, Perry G, Zhu X, Moreira PI, Smith MA. Insulin-resistant brain state: the culprit in sporadic Alzheimer’s disease? Ageing Res Rev (2011) 10(2):264–73. doi:10.1016/j.arr.2011.01.001
230. Baker LD, Cross DJ, Minoshima S, Belongia D, Watson GS, Craft S. Insulin resistance and Alzheimer-like reductions in regional cerebral glucose metabolism for cognitively normal adults with prediabetes or early type 2 diabetes. Arch Neurol (2011) 68(1):51–7. doi:10.1001/archneurol.2010.225
231. Biessels GJ, Staekenborg S, Brunner E, Brayne C, Scheltens P. Risk of dementia in diabetes mellitus: a systematic review. Lancet Neurol (2006) 5(1):64–74. doi:10.1016/S1474-4422(05)70284-2
232. Kuusisto J, Koivisto K, Mykkanen L, Helkala EL, Vanhanen M, Hanninen T, et al. Association between features of the insulin resistance syndrome and Alzheimer’s disease independently of apolipoprotein E4 phenotype: cross sectional population based study. BMJ (1997) 315(7115):1045–9. doi:10.1136/bmj.315.7115.1045
233. Matsuzaki T, Sasaki K, Tanizaki Y, Hata J, Fujimi K, Matsui Y, et al. Insulin resistance is associated with the pathology of Alzheimer disease: the Hisayama study. Neurology (2010) 75(9):764–70. doi:10.1212/WNL.0b013e3181eee25f
234. Tolppanen AM, Lavikainen P, Solomon A, Kivipelto M, Uusitupa M, Soininen H, et al. History of medically treated diabetes and risk of Alzheimer disease in a nationwide case-control study. Diabetes Care (2013) 36(7):2015–9. doi:10.2337/dc12-1287
235. Kim B, Backus C, Oh S, Hayes JM, Feldman EL. Increased tau phosphorylation and cleavage in mouse models of type 1 and type 2 diabetes. Endocrinology (2009) 150(12):5294–301. doi:10.1210/en.2009-0695
236. Ho L, Qin W, Pompl PN, Xiang Z, Wang J, Zhao Z, et al. Diet-induced insulin resistance promotes amyloidosis in a transgenic mouse model of Alzheimer’s disease. FASEB J (2004) 18(7):902–4. doi:10.1096/fj.03-0978fje
237. Papon MA, El Khoury NB, Marcouiller F, Julien C, Morin F, Bretteville A, et al. Deregulation of protein phosphatase 2A and hyperphosphorylation of tau protein following onset of diabetes in NOD mice. Diabetes (2013) 62(2):609–17. doi:10.2337/db12-0187
238. Planel E, Tatebayashi Y, Miyasaka T, Liu L, Wang L, Herman M, et al. Insulin dysfunction induces in vivo tau hyperphosphorylation through distinct mechanisms. J Neurosci (2007) 27(50):13635–48. doi:10.1523/JNEUROSCI.3949-07.2007
239. Takeda S. Pathological interaction between diabetes mellitus and Alzheimer’s disease. Nihon Shinkei Seishin Yakurigaku Zasshi (2012) 32(5–6):239–44.
240. Vandal M, White PJ, Tremblay C, St-Amour I, Chevrier G, Emond V, et al. Insulin reverses the high-fat diet-induced increase in brain abeta and improves memory in an animal model of Alzheimer disease. Diabetes (2014). doi:10.2337/db14-0375
241. Ott A, Stolk RP, van Harskamp F, Pols HA, Hofman A, Breteler MM. Diabetes mellitus and the risk of dementia: the Rotterdam study. Neurology (1999) 53(9):1937–42. doi:10.1212/WNL.53.9.1937
242. Cukierman T, Gerstein HC, Williamson JD. Cognitive decline and dementia in diabetes – systematic overview of prospective observational studies. Diabetologia (2005) 48(12):2460–9. doi:10.1007/s00125-005-0023-4
243. Schoenle EJ, Schoenle D, Molinari L, Largo RH. Impaired intellectual development in children with type I diabetes: association with HbA(1c), age at diagnosis and sex. Diabetologia (2002) 45(1):108–14. doi:10.1007/s125-002-8250-6
244. Dahlquist G, Kallen B. School performance in children with type 1 diabetes – a population-based register study. Diabetologia (2007) 50(5):957–64. doi:10.1007/s00125-007-0615-2
245. Musen G, Lyoo IK, Sparks CR, Weinger K, Hwang J, Ryan CM, et al. Effects of type 1 diabetes on gray matter density as measured by voxel-based morphometry. Diabetes (2006) 55(2):326–33. doi:10.2337/diabetes.55.02.06.db05-0520
246. Steen E, Terry BM, Rivera EJ, Cannon JL, Neely TR, Tavares R, et al. Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer’s disease – is this type 3 diabetes? J Alzheimers Dis (2005) 7(1):63–80.
247. Serbedzija P, Ishii DN. Insulin and insulin-like growth factor prevent brain atrophy and cognitive impairment in diabetic rats. Indian J Endocrinol Metab (2012) 16(Suppl 3):S601–10. doi:10.4103/2230-8210.105578
248. MacKnight C, Rockwood K, Awalt E, McDowell I. Diabetes mellitus and the risk of dementia, Alzheimer’s disease and vascular cognitive impairment in the Canadian study of health and aging. Dement Geriatr Cogn Disord (2002) 14(2):77–83. doi:10.1159/000064928
249. Hassing LB, Johansson B, Nilsson SE, Berg S, Pedersen NL, Gatz M, et al. Diabetes mellitus is a risk factor for vascular dementia, but not for Alzheimer’s disease: a population-based study of the oldest old. Int Psychogeriatr (2002) 14(3):239–48. doi:10.1017/S104161020200844X
250. de la Monte SM, Wands JR. Molecular indices of oxidative stress and mitochondrial dysfunction occur early and often progress with severity of Alzheimer’s disease. J Alzheimers Dis (2006) 9(2):167–81.
251. Mosconi L. Brain glucose metabolism in the early and specific diagnosis of Alzheimer’s disease. FDG-PET studies in MCI and AD. Eur J Nucl Med Mol Imaging (2005) 32(4):486–510. doi:10.1007/s00259-005-1762-7
252. Minoshima S, Giordani B, Berent S, Frey KA, Foster NL, Kuhl DE. Metabolic reduction in the posterior cingulate cortex in very early Alzheimer’s disease. Ann Neurol (1997) 42(1):85–94. doi:10.1002/ana.410420114
253. Simpson IA, Chundu KR, Davies-Hill T, Honer WG, Davies P. Decreased concentrations of GLUT1 and GLUT3 glucose transporters in the brains of patients with Alzheimer’s disease. Ann Neurol (1994) 35(5):546–51. doi:10.1002/ana.410350507
254. Liu F, Shi J, Tanimukai H, Gu J, Grundke-Iqbal I, Iqbal K, et al. Reduced O-GlcNAcylation links lower brain glucose metabolism and tau pathology in Alzheimer’s disease. Brain (2009) 132(Pt 7):1820–32. doi:10.1093/brain/awp099
255. Mastrogiacoma F, Bettendorff L, Grisar T, Kish SJ. Brain thiamine, its phosphate esters, and its metabolizing enzymes in Alzheimer’s disease. Ann Neurol (1996) 39(5):585–91. doi:10.1002/ana.410390507
256. Brunham LR, Kruit JK, Verchere CB, Hayden MR. Cholesterol in islet dysfunction and type 2 diabetes. J Clin Invest(2008) 118(2):403–8. doi:10.1172/JCI33296
257. Osborne AR, Pollock VV, Lagor WR, Ness GC. Identification of insulin-responsive regions in the HMG-CoA reductase promoter. Biochem Biophys Res Commun (2004) 318(4):814–8. doi:10.1016/j.bbrc.2004.04.105
258. Nelson TJ, Alkon DL. Insulin and cholesterol pathways in neuronal function, memory and neurodegeneration. Biochem Soc Trans (2005) 33(Pt 5):1033–6. doi:10.1042/BST20051033
259. Nelson TJ, Alkon DL. Oxidation of cholesterol by amyloid precursor protein and beta-amyloid peptide. J Biol Chem(2005) 280(8):7377–87. doi:10.1074/jbc.M409071200
260. Liu F, Iqbal K, Grundke-Iqbal I, Hart GW, Gong CX. O-GlcNAcylation regulates phosphorylation of tau: a mechanism involved in Alzheimer’s disease. Proc Natl Acad Sci U S A (2004) 101(29):10804–9. doi:10.1073/pnas.0400348101
261. Dias WB, Hart GW. O-GlcNAc modification in diabetes and Alzheimer’s disease. Mol Biosyst (2007) 3(11):766–72. doi:10.1039/b704905f
262. Liu Y, Liu F, Grundke-Iqbal I, Iqbal K, Gong CX. Brain glucose transporters, O-GlcNAcylation and phosphorylation of tau in diabetes and Alzheimer’s disease. J Neurochem (2009) 111(1):242–9. doi:10.1111/j.1471-4159.2009.06320.x
263. Finder VH, Glockshuber R. Amyloid-beta aggregation. Neurodegener Dis (2007) 4(1):13–27. doi:10.1159/000100355
264. Li ZG, Zhang W, Sima AA. Alzheimer-like changes in rat models of spontaneous diabetes. Diabetes (2007) 56(7):1817–24. doi:10.2337/db07-0171
265. Zhao L, Teter B, Morihara T, Lim GP, Ambegaokar SS, Ubeda OJ, et al. Insulin-degrading enzyme as a downstream target of insulin receptor signaling cascade: implications for Alzheimer’s disease intervention. J Neurosci (2004) 24(49):11120–6. doi:10.1523/JNEUROSCI.2860-04.2004
266. Farris W, Mansourian S, Chang Y, Lindsley L, Eckman EA, Frosch MP, et al. Insulin-degrading enzyme regulates the levels of insulin, amyloid beta-protein, and the beta-amyloid precursor protein intracellular domain in vivo. Proc Natl Acad Sci U S A (2003) 100(7):4162–7. doi:10.1073/pnas.0230450100
267. Johnson GV. Tau phosphorylation and proteolysis: insights and perspectives. J Alzheimers Dis (2006) 9(3 Suppl):243–50.
268. Rissman RA, Poon WW, Blurton-Jones M, Oddo S, Torp R, Vitek MP, et al. Caspase-cleavage of tau is an early event in Alzheimer disease tangle pathology. J Clin Invest (2004) 114(1):121–30. doi:10.1172/JCI20640
269. Holmes C, Cotterell D. Role of infection in the pathogenesis of Alzheimer’s disease: implications for treatment. CNS Drugs (2009) 23(12):993–1002. doi:10.2165/11310910-000000000-00000
270. Alonso R, Pisa D, Marina AI, Morato E, Rabano A, Carrasco L. Fungal infection in patients with Alzheimer’s disease. J Alzheimers Dis (2014) 41(1):301–11. doi:10.3233/JAD-132681
271. Alonso R, Pisa D, Rabano A, Carrasco L. Alzheimer’s disease and disseminated mycoses. Eur J Clin Microbiol Infect Dis(2014) 33(7):1125–32. doi:10.1007/s10096-013-2045-z
272. Asai M, Hattori C, Iwata N, Saido TC, Sasagawa N, Szabo B, et al. The novel beta-secretase inhibitor KMI-429 reduces amyloid beta peptide production in amyloid precursor protein transgenic and wild-type mice. J Neurochem (2006) 96(2):533–40. doi:10.1111/j.1471-4159.2005.03576.x
273. Noble W, Planel E, Zehr C, Olm V, Meyerson J, Suleman F, et al. Inhibition of glycogen synthase kinase-3 by lithium correlates with reduced tauopathy and degeneration in vivo. Proc Natl Acad Sci U S A (2005) 102(19):6990–5. doi:10.1073/pnas.0500466102
274. Blass JP, Gleason P, Brush D, DiPonte P, Thaler H. Thiamine and Alzheimer’s disease. A pilot study. Arch Neurol(1988) 45(8):833–5. doi:10.1001/archneur.1988.00520320019008
275. Nolan KA, Black RS, Sheu KF, Langberg J, Blass JP. A trial of thiamine in Alzheimer’s disease. Arch Neurol (1991) 48(1):81–3. doi:10.1001/archneur.1991.00530130093025
276. Radhakrishnan ML, Tidor B. Optimal drug cocktail design: methods for targeting molecular ensembles and insights from theoretical model systems. J Chem Inf Model (2008) 48(5):1055–73. doi:10.1021/ci700452r
277. Kickstein E, Krauss S, Thornhill P, Rutschow D, Zeller R, Sharkey J, et al. Biguanide metformin acts on tau phosphorylation via mTOR/protein phosphatase 2A (PP2A) signaling. Proc Natl Acad Sci U S A (2010) 107(50):21830–5. doi:10.1073/pnas.0912793107
278. Hsu CC, Wahlqvist ML, Lee MS, Tsai HN. Incidence of dementia is increased in type 2 diabetes and reduced by the use of sulfonylureas and metformin. J Alzheimers Dis (2011) 24(3):485–93. doi:10.3233/JAD-2011-101524
279. Kintscher U, Law RE. PPARgamma-mediated insulin sensitization: the importance of fat versus muscle. Am J Physiol Endocrinol Metab (2005) 288(2):E287–91. doi:10.1152/ajpendo.00440.2004
280. Combs CK, Johnson DE, Karlo JC, Cannady SB, Landreth GE. Inflammatory mechanisms in Alzheimer’s disease: inhibition of beta-amyloid-stimulated proinflammatory responses and neurotoxicity by PPARgamma agonists. J Neurosci (2000) 20(2):558–67.
281. Delerive P, Fruchart JC, Staels B. Peroxisome proliferator-activated receptors in inflammation control. J Endocrinol(2001) 169(3):453–9. doi:10.1677/joe.0.1690453
282. Watson GS, Cholerton BA, Reger MA, Baker LD, Plymate SR, Asthana S, et al. Preserved cognition in patients with early Alzheimer disease and amnestic mild cognitive impairment during treatment with rosiglitazone: a preliminary study. Am J Geriatr Psychiatry (2005) 13(11):950–8. doi:10.1176/appi.ajgp.13.11.950
283. McClean PL, Parthsarathy V, Faivre E, Holscher C. The diabetes drug liraglutide prevents degenerative processes in a mouse model of Alzheimer’s disease. J Neurosci (2011) 31(17):6587–94. doi:10.1523/JNEUROSCI.0529-11.2011
284. Holscher C. The role of GLP-1 in neuronal activity and neurodegeneration. Vitam Horm (2010) 84:331–54. doi:10.1016/B978-0-12-381517-0.00013-8
285. Holscher C. Incretin analogues that have been developed to treat type 2 diabetes hold promise as a novel treatment strategy for Alzheimer’s disease. Recent Pat CNS Drug Discov (2010) 5(2):109–17. doi:10.2174/157488910791213130
286. Li Y, Duffy KB, Ottinger MA, Ray B, Bailey JA, Holloway HW, et al. GLP-1 receptor stimulation reduces amyloid-beta peptide accumulation and cytotoxicity in cellular and animal models of Alzheimer’s disease. J Alzheimers Dis (2010) 19(4):1205–19. doi:10.3233/JAD-2010-1314
287. Hamilton A, Patterson S, Porter D, Gault VA, Holscher C. Novel GLP-1 mimetics developed to treat type 2 diabetes promote progenitor cell proliferation in the brain. J Neurosci Res (2011) 89(4):481–9. doi:10.1002/jnr.22565
288. Craft S. Insulin resistance syndrome and Alzheimer’s disease: age- and obesity-related effects on memory, amyloid, and inflammation. Neurobiol Aging (2005) 26(Suppl 1):65–9. doi:10.1016/j.neurobiolaging.2005.08.021
289. Craft S, Baker LD, Montine TJ, Minoshima S, Watson GS, Claxton A, et al. Intranasal insulin therapy for Alzheimer disease and amnestic mild cognitive impairment: a pilot clinical trial. Arch Neurol (2012) 69(1):29–38. doi:10.1001/archneurol.2011.233
290. Ott V, Benedict C, Schultes B, Born J, Hallschmid M. Intranasal administration of insulin to the brain impacts cognitive function and peripheral metabolism. Diabetes Obes Metab (2012) 14(3):214–21. doi:10.1111/j.1463-1326.2011.01490.x
291. Nakazato M. Development of the novel delivery system of GLP-1 administration for the treatment of diabetes mellitus. Nihon Rinsho (2011) 69(5):918–22.
292. Copps KD, White MF. Regulation of insulin sensitivity by serine/threonine phosphorylation of insulin receptor substrate proteins IRS1 and IRS2. Diabetologia (2012) 55(10):2565–82. doi:10.1007/s00125-012-2644-8
Купить номер с этой статьей в pdf