Болезнь Альцгеймера как одно из проявлений сахарного диабета 3 типа
Nguyen TT, Ta QTH, Nguyen TKO, Nguyen TTD, Giau VV
Перевод из журнала International Journal of Molecular Sciences (оригинал).
Аннотация
Точная связь между болезнью Альцгеймера и сахарным диабетом 2 типа все еще не установлена, однако достоверно известно, что при длительном повышении уровня глюкозы в крови риск развития данного нейродегенеративного заболевания увеличивается.
Учитывая рост количества данных по этой проблеме, обзор призван продемонстрировать взаимосвязь между нарушением обмена глюкозы в центральной нервной системе, что обозначается как сахарный диабет 3 типа, и развитием болезни Альцгеймера на основании того факта, что токсичность белков-предшественников амилоида ассоциирована с нарушением передачи сигналов от инсулина периферическим тканям, то есть с развитием инсулинорезистентности. Указанные изменения служат причиной нарушения многих биохимических процессов в нервных клетках и прогрессированию болезни Альцгеймера.
Предполагается, что терапевтические стратегии, связанные с воздействием на уровень инсулина, могут быть эффективны при болезни Альцгеймера вследствие замедления прогрессирования заболевания при нормализации уровня глюкозы в центральной нервной системе.
Введение
Сахарный диабет (СД) представляет собой тяжелое хроническое заболевание, оказывающее серьезное влияние на жизнь и благополучие как индивидов, так и семей, и общества в целом. Распространенность диабета в мире в 2019 году оценивается в 9,3% (463 миллиона человек), и потенциально возрастет до 10,2% (578 миллионов) к 2030 году и до 10,9% (700 миллионов) к 2045 году [1].
Старение населения планеты резко увеличивается, особенно в развитых странах, создавая нагрузку на систему здравоохранения, а также на службы социального обеспечения. С увеличением распространенности СД около 5,76 миллионов человек, страдающих этим заболеванием, в настоящее время проживает во Вьетнаме. Согласно прогнозам, СД станет во Вьетнаме к 2030 г одним из семи основных заболеваний, ведущих к инвалидности и смерти [2,3].
В настоящее время многие люди осведомлены, что существует СД 1 и 2 типа, однако недавно ученые предложили выделить еще одну форму диабета, известную как диабет 3 типа (СД 3 типа). Этот менее известный тип заболевания проявляется как инсулинорезистентность тканей головного мозга, что влияет на когнитивные функции и вносит свой вклад в развитие болезни Альцгеймера (БА) [4].
СД 1 типа в основном возникает из-за разрушения β-клеток, что приводит к абсолютной недостаточности инсулина. СД 2 типа развивается вследствие прогрессирующего снижения секреции инсулина одновременно с резистентностью к этому гормону. Инсулинорезистентность - распространенное явление, тесно связанное с ожирением и определяемое как неспособность тканей-мишеней адекватно воспринимать инсулин. Обычно это состояние предшествует возникновение СД 2 типа.
СД 1 типа в основном наблюдается у детей и молодых людей, тогда как СД 2 типа чаще встречается у взрослых и является причиной 90% случаев нарушения обмена глюкозы во всем мире [5,6].
Недавно открытая учеными форма СД получила название сахарного диабета 3 типа (СД 3 типа). Исследователи предложили рассматривать данное заболевание как метаболический синдром, который может привести к прогрессирующей инсулинорезистентности ткани головного мозга с последующим нарушением процессов передачи сигналов посредством инсулина, накоплением нейротоксинов, развитием нейронального стресса и в итоге нейродегенерации [7,8].
БА служит шестой по частоте среди ведущих причин смерти в США и пятой по значимости – среди людей 65 лет и старше. Этиотропного лечения от этого заболевания в настоящий момент не разработано, возможна лишь симптоматическая терапия.
Дефицит нейротрансмиттеров, дегенерация нейронов, дисфункция синапсов, накопление ß-амилоида и внутриклеточные нейрофибриллярные сплетения являются основными патоморфологическими изменениями со стороны ткани мозга при БА [5].
В настоящий момент ученые признают, что СД 3 типа и БА имеют схожие механизмы развития и факторы риска, в большей степени это условия окружающей среды, в меньшей – генетическая предрасположенность.
Исследования in vitro и на животных показали, что инсулинорезистентность может вносить вклад в патогенез БА посредством множества различных путей [7]. СД способен влиять на обработку данных в памяти (распознавание и извлечение), морфологию мозга и синаптическую связь. Указанные нарушения наблюдаются при БА [9]. Кроме того, гиперинсулинемия, нарушение передачи сигналов от инсулина к тканям и резистентность к этому гормону служат жизненно важными факторами, которые определяют центральное место обмена инсулина в организме при рассмотрении указанных двух заболеваний независимо от генотипа [10].
Многие исследования продемонстрировали снижение памяти и ухудшение когнитивных способностей из-за нарушения передачи сигналов от инсулина к тканям гиппокампа [11–13]. Периферическая инсулинорезистентность приводит к снижению скорости передачи сигналов от инсулина в ЦНС, далее следует изменение метаболизма мозга. Указанный факт объясняет тесную связь между гиперинсулинемией, инсулинорезистентностью и возникающими в результате заболеваниями, такими как СД 3 типа и БА [14].
Повышенная токсичность Aß- белков, окислительный стресс, гиперфосфорилирование тау-белка и нейровоспаление также имеют ассоциацию с резистентностью к инсулину тканей мозга, что в итоге приводит к нейродегенерации (Рисунок 1).
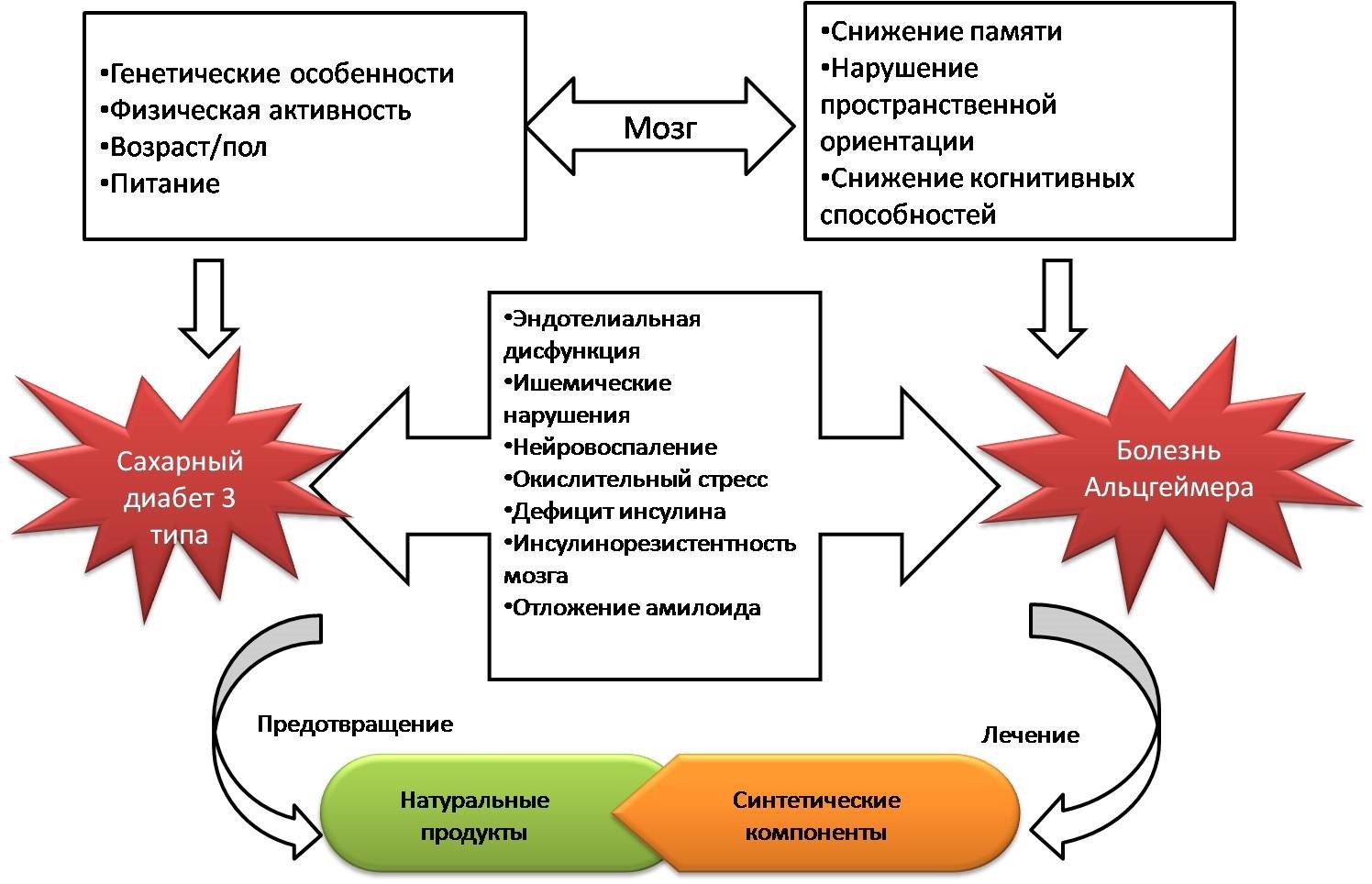
Рисунок 1. Связь СД 3 типа и БА, подходы к лечению этих заболеваний
Передача сигналов инсулина в центральной нервной системе
Инсулин – это гормон, регулирующий уровень глюкозы в крови, за продукцию которого отвечают бета-клетки так называемых островков Лангерганса в поджелудочной железе. Данный гормон состоит из двух полипептидных цепей, соединенных с помощью дисульфидных связей. Инсулин инициирует свое действие путем связывания с рецепторами трансмембранных гликопротеинов, состоящих из двух ?- и двух ß- субъединиц [14]. Связывание инсулина с β-субъединицами рецепторов приводит к аутофосфорилированию нескольких остатков аминокислоты тирозина в ß-цитозольной области субъединицы [15,16].
Аутофосфорилированные остатки затем распознаются субстратами рецепторов инсулина (IRS), из которых IRS-1 и IRS-2 являются двумя основными участниками и посредниками в распространении сигнала от инсулина. IRS подходит под конфигурацию молекулярных комплексов, опосредующих внутриклеточные сигнальные пути. Инсулин и инсулиноподобный фактор роста -1 (IGF-1) соединяются с соответствующими рецепторами. Соединение этих рецепторов с инсулином наиболее высоко в обонятельной луковице, коре головного мозга и гиппокампе. Кроме того, рецепторы инсулина также экспрессируются на поверхности эндотелиальных клеток гематоэнцефалического барьера (ГЭБ) и отвечают за транспорт инсулина и IGF-1 через этот барьер в ЦНС [17].
Несмотря на то, что точный механизм проникновения инсулина в мозг все еще остается спорным, предполагается, что инсулин, циркулирующий в крови, может проникать сквозь ГЭБ через рецептор-опосредованную активную транспортную систему [17]. Это предположение согласуется с исследованиями, в которых было обнаружено, что уровень инсулина в спинномозговой жидкости увеличивается пропорционально таковому в крови после инфузии [15–17].
Существует также предположение о некотором количестве инсулина, вырабатываемом в ЦНС, однако, насколько это значимо, остается неясным. Возможно, что оба способа поступления инсулина, как из центра, так и с периферии, важны для передачи сигналов в головном мозге.
Инсулин и IGF-1 наделены функциями, которые важны для выживания нейронов и поддержания гомеостаза в ЦНС. Механизмы действия данных молекул связаны с поступлением в клетки кальция, образованием нейротрансмиттеров и синаптических связей, регуляцией апоптоза и нейрогенеза [17]. Инсулин также регулирует экспрессию рецепторов гамма-аминомасляной кислоты, играет решающую роль в увеличении числа синапсов и формировании дендритных шипиков за счет активации AKT – mTOR - и Ras-связанных путей, которые служат неотъемлемой частью передачи сигналов инсулина [18 - 21]. Также данный гормон влияет на выживаемость клеток путем регуляции образования промежуточных соединений, участвующих в апоптозе [22,23].
О наличии инсулина в головном мозге впервые сообщили Havrankova et al, которые применили радиоиммуноанализ для определения уровня инсулина в ткани мозга. [24] Высокие концентрации инсулина были обнаружены не только в мозге экспериментальных животных, но и человека [25]. В последнее время синтез инсулина в ЦНС также широко изучается. Доказано присутствие мРНК инсулина в перивентрикулярном ядре гипоталамуса крыс методом гибридизации in situ [26].
Молекулярные механизмы, опосредующие производство и секрецию инсулина в ЦНС, обнаруживают сходство между ß-клетками поджелудочной железы и нейронами, особенно в отношении АТФ-чувствительных калиевых каналов [27]. Указанное высвобождение инсулина, вызванное деполяризацией нейронов, в экспериментах демонстрировало подавление циклогексимидом и было специфичным для нейронов, но не для астроцитов [28].
Интересно, что нарушение процессов, опосредованных инсулиновыми рецепторами, может быть связано с патологией активации этих рецепторов и снижением доступности инсулина, что в итоге приводит к широкому спектру нарушений работы мозга [29, 30].
Таким образом, инсулин обладает возможностью влиять на производительность нейронов и их целостность, а его дефицит в тканях мозга приводит к когнитивным дефектам, снижению памяти и другим проблемам, связанным с БА. Тем не менее, требуется больше данных для улучшения понимания функций инсулина в ЦНС и его влиянием на развитие нейродегенеративных изменений мозга [31].
Роль гомеостаза глюкозы в развитии сахарного диабета 3 типа
Ключ к пониманию взаимосвязи между СД и нейродегенеративными изменениями лежит в особенностях энергетических процессов в головном мозге при диабете. Энергетический гомеостаз представляет собой хорошо регулируемый процесс, который зависит от согласованности потребления и расхода энергии. Поддержанию гомеостаза энергии у людей в последние годы уделялось много внимания в исследованиях из-за роста заболеваемости ожирением и диабетом.
Обнаружено, что взрослые нейроны имеют две отличительные особенности, которые делают их подверженными гибели или патологическим изменениям, таким как нейродегенерация. Первая особенность состоит в том, что полностью дифференцированные нейроны лишены регенерационной способности [32]. Следовательно, когда они подвергаются каким-либо негативным воздействиям, то либо сразу погибают, либо подвергаются апоптозу, что служит предпосылкой для развития нейродегенеративных заболеваний [32]. Второй важной особенностью служит особая требовательность нейронов и ткани мозга в целом к энергетическим ресурсам. Так, более 40% присутствующего АТФ используется для поддержания жизнеспособности нейронов [33].
Существует два источника глюкозы в головном мозге, которые стимулируют метаболизм в корковом веществе: базальный уровень инсулина крови и превращение астроцитарного гликогена в глюкозу [34]. Увеличение захвата глюкозы осуществляется с помощью инсулин-чувствительного глиального транспортера глюкозы типа 1 (GLUT1), который доставляет ее к плазматической мембране с последующим использованием нейронами. Следовательно, сбалансированный клеточный транспорт глюкозы зависит от астроцитов и переносчиков глюкозы, которые экспрессируются в головном мозге [35].
Нарушение метаболизма глюкозы в ЦНС может быть важным фактором в патогенезе СД 3 типа. Механизмы, которые вовлечены в нарушения транспорта глюкозы, включают инсулинорезистентность тканей головного мозга и изменение внутриклеточного обмена глюкозы.
Уменьшение количества переносчиков глюкозы продемонстрировало в исследованиях корреляцию с патологическим гиперфосфорилированием тау-белков при нейродегенеративных заболеваниях [36]. Следовательно, нарушение передачи сигналов от инсулина влияет не только на уровень глюкозы в крови, но также способствует развитию дегенеративных процессов в клетках мозга и гибели нейронов [37].
Кроме того, инсулинорезистентность при СД 2 типа определяется как «снижение чувствительности тканей организма к действию инсулина» [38]. Точно также инсулинорезистентность мозга можно определить как неспособность клеток мозга адекватно реагировать на сигналы инсулина [39]. Следовательно, это состояние приводит к дефициту инсулина и нарушению транспорта глюкозы внутрь нейронов из-за уменьшения количества экспрессии GLUT в мембране.
Отсутствие ответа на действие инсулина может повышать чувствительность нейронов к токсическим воздействиям [40]. Значительное увеличение при этом таких патологических признаков, как апоптоз и нейродегенерация, служит предпосылкой для снижения когнитивных функций. [41].
Таким образом, нарушение гомеостаза глюкозы играет важную роль в патогенезе нейродегенеративных заболеваний. Следовательно, сочетание СД 2 типа и БА можно рассматривать в качестве нового типа нейроэндокринного заболевания – СД 3 типа.
Сахарный диабет 3 типа и накопление Аß-белков в головном мозге
Основой амилоидоза служит патологическое накопление фибриллярных белков - амилоидов вне клеток. Клинические проявления этого заболевания различны в зависимости от пораженного органа. Согласно новым данным, СД 2 типа можно рассматривать в качестве фактора риска для образования отложений ß-амилоида в головном мозге у пациентов с деменцией.
Кроме того, имеется связь между длительным непрерывным воздействием инсулина на ткань мозга и накоплением Аß-белков внутри нейронов [42]. Согласно Farris et al., фермент, разрушающий инсулин (IDE), регулирует также количество Аß-белков и белка-предшественника амилоида in vivo [42]. Это исследование показало, что СД 2 типа у крыс с мутантным вариантом IDE был связан с гиперинсулинемией и нарушением метаболизма глюкозы в головном мозге.
Снижение функциональной активности IDE, таким образом, может играть роль в развитии СД 3 типа, способствуя дегенерации и гибели нейронов [42]. У здоровых субъектов IDE снижает уровень Аß-белков, регулирует действие инсулина, а также разрушает внутриклеточный домен белка-предшественника амилоида (AICD). В случае инсулинорезистентности мозга этот механизм нарушается, и в ЦНС образуются патологические скопления белков [42].
В отношении СД 3 типа и инсулинорезистентности головного мозга ведутся споры, является ли нарушение обмена глюкозы следствием или причиной аномальной экспрессии и процессинга Аß-белков [43]. Если рассматривать СД 3 типа как следствие, то первичной представляется токсическое действие указанных белков на нейроны, а именно нарушение передачи сигналов от инсулина в клетку, что вызывает их инсулинорезистентность [44, 45].
С другой стороны, возможна и обратная ситуация, то есть нарушение метаболизма глюкозы служит причиной нейровоспаления и формирования патологических отложений белков. Исследования, основанные на этой концепции, утверждают, что стимуляция инсулином может увеличивать или ускорять движение Аß-белков из сети Гольджи до плазматической мембраны. Следовательно, инсулин способен активировать экскрецию патологических белков из клетки и в то же время ингибировать их внутриклеточное накопление [46]. Таким образом, нарушение передачи сигналов инсулина уменьшает клиренс указанных белков, то есть способствует их скоплению и нейротоксическому действию на клетки нервной системы [47] (Рисунок 2).
Рисунок 2. Схема общих метаболических путей инсулинорезистентности и болезни Альцгеймера
Интересен тот факт, что пациенты с СД 2 типа и БА имеют аналогичные отложения бета-амилоида как в поджелудочной железе, так и в головном мозге. Это служит еще одной причиной, почему данные заболевания некоторые ученые рассматривают в общей парадигме СД 3 типа [48–51].
Связь сахарного диабета 3 типа с болезнью Альцгеймера
В последнее время многие исследования продемонстрировали повышение частоты БА у пациентов с СД 2 типа и лиц с ожирением, что подразумевает общие механизмы развития этих заболеваний [10,52,53]. Основным признаком, общим для диабета, ожирения и БА, служит инсулинорезистентность [54].
Утилизация глюкозы нейронами может не полностью зависеть от уровня инсулина, влияние инсулинорезистентности на мозг также связано с нарушением сигнальных путей данного гормона [55]. Инсулинорезистентость способствует нарушению функций нейронов, что сопровождается экстремальным повышение уровня инсулина [56,57]. Это замедляет цереброкортикальный метаболизм глюкозы, и, как следствие, нарушает проведение нервных импульсов в гиппокамп, снижая когнитивные функции и память [50].
Предыдущие исследования продемонстрировали, что пациенты с продромальными явлениями БА имеют повышенный уровень рецепторов IRS1, что свидетельствует о развитии инсулинорезистентности при БА за несколько лет до клинических проявлений заболевания [58].
Из-за нарушения действия инсулина отмечается неправильная активация передачи сигналов от рецепторов к тканям мозга. Основным последствием этого патологического каскада реакций служит снижение утилизации глюкозы нейронами, которое проявляется нарушением нейропластичности, дефицитом нейротрансмиттеров и снижением биоэнергетического потенциала клеток.
Общие представления о взаимосвязи процессов, характерных для СД 3 типа и БА, выделены в таблице 1.
| Факторы риска | Метаболические предшественники | Механизмы | Субклиническая патология | Исход | |
| Социальные факторы: стресс, низкий социально-экономический статус, определенная этническая и расовая группа |
Ожирение, в том числе висцеральное | Сосудистые процессы: Артериальная гипертензия
Гиперлипидемия
Аполипопротеин E |
Церебральный кровоток
Атеросклероз |
Белки-предшественники амилоида |
БА |
| Плохое питание: употребление большого количества калорий, жиров и сахара | Воспалительные / окислительные процессы:
Воспаление Окислительный стресс Эндотелиальная функция |
Нейрофибриллярные отложения амилоида
| |||
| Физическое бездействие Генетика и история семьи | Гипергликемия Гиперинсулинемия | Метаболические процессы: Инсулинорезистентность Фермент, расщепляющий инсулин Рецепторы, активирующиеся пролиферацией пероксисом
|
| ||
| Раннее детство воздействия в утробе матери и вес при рождении |
| Атрофия головного мозга и гиппокампа Увеличение интенсивности отображения белого вещества |
| ||
Таблица 1. Модель, объясняющая взаимосвязь сахарного диабета 3 типа и болезни Альцгеймера
СД 3 типа возникает, когда нейроны в головном мозге теряют способность реагировать на инсулиновые сигналы, что необходимо для осуществления основных задач мозга, включая запоминание и обучение. Некоторые исследователи считают, что дефицит инсулина играет ключевую роль в снижении когнитивных функций при БА.
На молекулярном уровне клетка взаимодействует с инсулином через соответствующие рецепторы, при этом сигнал распространяется с помощью каскада реакций, известных под общим названием "сигнальный путь PI3K / Akt / mTOR". Чувствительность этого пути к инсулину может быть снижена ввиду воздействия многих факторов, вызывающих инсулинорезистентность. На рисунке 3 представлено схематическое отображение связи инсулинорезистентности тканей головного мозга с развитием БА.
Рисунок 3. Связь инсулинорезистентности головного мозга, накопления Аß-белков и их токсического воздействия на центральную нервную систему
Недавние научные работы показали, что ответ клетки на действие инсулина может быть пороговым явлением [13,59,60]. Сопротивление к влиянию инсулина может быть быстро преодолено путем воздействия на митохондриальные разобщители клетки и некоторые другие механизмы [61,62].
В некоторых клинических исследованиях сообщается о снижении толерантности к глюкозе у пациентов с БА и предполагается двухсторонняя связь между сахарным диабетом и БА [63,64]. В экспериментах на животных у десятимесячных мышей наблюдалось снижение уровня IRS-1 в гиппокампе и коре головного мозга [65, 66]. Маркеры инсулинорезистентности также обнаружены в гипоталамусе и фронтальной коре головного мозга животных [67-70]. Чтобы подтвердить данную концепцию, необходимы дальнейшие исследования механизмов, посредством которых БА влияет на диабетический фенотип.
Инсулин регулирует гомеостаз глюкозы и липидов за счет передачи сигналов в клетки печени, скелетных мышц и жировой ткани. Нарушение передачи сигналов от инсулина и инсулиноподобного фактора роста-1 (IGF1) является фактором риска когнитивных нарушений и деменции, включая БА [71]. Системная гетерозиготная инактивация IGF1R (IGF1R + /-) или нейрональная делеция IGF1R (nIGF1R- /-) улучшает выживаемость в мышиной модели БА при одновременном снижении поведенческих нарушений и уменьшении накоплений Аß-белков [72]. Снижение передачи сигналов IRS2 в мозге продлевает жизнь, улучшает когнитивные функции и уменьшает количество отложений Аß-белков у мышей Tg2576 с нормальным уровнем глюкозы в крови [72-74].
Терапевтические подходы к лечению сахарного диабета 3 типа и болезни Альцгеймера
Инсулинорезистентность хорошо известна как неотъемлемая черта СД 3 типа, поэтому стратегии лечения, направленные на повышение чувствительности к инсулину, могут принести пользу пациентам в снижении риска развития БА на ранних стадиях. Совпадение патогенетических механизмов развития СД 3 типа, инсулинорезистентности и нейродегенерации привело к поиску многоцелевых лекарственных средств, среди которых предложены антиоксиданты, полифенолы, жирные кислоты омега-3, препараты, влияющие на ось "кишечник-мозг" [75-77].
Среди указанных терапевтических агентов в исследованиях присутствует куркумин. Это вещество продемонстрировало возможность уменьшения объема патологических скоплений белков, способность препятствовать проапоптотическим сигнальным путям в нейронах гиппокампа [78].
Предыдущие исследования также отметили пользу метформина у мышей в сочетании с добавлением куркумина и пиперина, особенно в отношении снижения инсулинорезистентности [78].
Хорошо известны противовоспалительные свойства фруктов и овощей, их способность противостоять окислительному стрессу и уменьшать повреждения клеток [79]. Исследования на модели мышей демонстрируют связь употребления овощей и фруктов с защитным эффектом против снижения когнитивных способностей ввиду наличия в их составе множества биоактивных компонентов, таких как каротиноиды, витамины, полифенолы и флавоноиды [80]. Различные семейства флавоноидов были предложены в качестве потенциальных терапевтических агентов после изучения их действия на модели животных [81].
Влияние омега-3 жирных кислот на развитие мозга и поддержание его функций получило широкое признание, особенно в последние десятилетия, но лишь недавно была продемонстрирована их роль в замедлении старения мозга [82]. Диета, богатая омега-3 жирными кислотами, может иметь ключевое значение для нутритивной терапии пациентов с БА [83].
Кетогенная диета служит спорным методом улучшения здоровья, однако получены некоторые данные об уменьшении амилоидных отложений в мозге одновременно с восстановлением поврежденных митохондрий и уменьшением воспаления на фоне питания продуктами, входящими в ее состав [84].
Новое исследование продемонстрировало, что гликирование белка APOE4 и неправильная передача сигналов инсулина приводит к нарушению транспорта липидов в ткани мозга [84-86]. Изменения гена APOE и периферическая инсулинорезистентность, вызванная диетой с высоким содержанием жиров, способствуют развитию изменений в головном мозге [87]. Белок APOE4 может более плотно связываться с рецепторами инсулина на поверхности нейронов, чем его нормальный аналог APOE3, и являться токсичным для нервных клеток [87].
В настоящее время не существует более эффективного способа для улучшения васкуляризации органов, в том числе мозга, чем физические упражнения. [88]. Это актуально и для людей, страдающих СД и БА. Достаточная физическая активность повышает качество жизни, способствует улучшению нейрохимического обмена в мозге, снижению инсулинорезистентности и ускорению клиренса белков - предшественников амилоида.
Терапевтические агенты, продемонстрировавшие эффективность в лечении патологии мозга, связанной с СД 3 типа и БА, указаны в таблице 2.
| Действующее вещество | Эффект лечения Реклама | Дизайн исследования | Ссылка |
| DA5-CH | Уменьшает фосфорилирование тау-белка
| Введение стрептозоцина крысам интрацеребровентрикулярно | [89] |
| DA-JC1 | Антагонист нарушения циркадных ритмов из-за накопления Аß-белков | Введение интрацеребровентрикулярно животным с БА | [90] |
| DA5-CH | Улучшение пластичности синапсов гиппокампа и активация PI3K / AKT сигнального пути | APP/PS1 мышиная модель БА model of AD | [91] |
| DA-CH3 | Снижение стресса и передачи сигналов, запускающих апоптоз нейронов, снижение образования амилоидных бляшек в головном мозге | APP/PS1 мышиная модель БА model of AD | [92] |
| Инсулин | Предотвращение индуцированной Аß -олигомером потери синапсов и уменьшения рецепторов инсулина | Культура клеток гиппокампа крыс | [93,94] |
| Инсулин | Пациенты с БА, имеющие Пониженную чувствительность к инсулину
| Пациенты с БА, гомозиготные или нет по ApoE 4, контрольная группа, введение внутривенно | [95] |
| Инсулин | Улучшение вербальной памяти после введения инсулина у пациентов с легкими когнитивными нарушениями | Пациенты с БА и легкими когнитивными нарушениями, гомозиготные или нет по ApoE 4, введение интраназально | [96,97] |
| Инсулин | Увеличение дозы интраназального инсулина улучшило выборочное внимание, удержание новой информации у пациентов с легкими когнитивными нарушениями и ранними признаками БА | Пациенты с БА, с легкими когнитивными нарушениями и здоровые люди, введение интраназально | [98] |
| Инсулин | Только среди женщин отмечено улучшение памяти после лечения Реклама | Здоровые мужчины и женщины, введение интраназально | [99] |
| Лираглутид | Уменьшение фосфорилирования тау-белка, защитное воздействие на рецепторы инсулина и синапсы | Введение макакам-крабоедам интрацеребровентрикулярно с Аß -олигомером
| [100] |
| Лираглутид | Улучшение памяти при распознании новых объектов или в опасной ситуации | Введение мышам интрацеребровентрикулярно с Аß -олигомером
| [100] |
| Лираглутид | Восстановление памяти при выполнении теста распознавания и прохождении водного лабиринта Морриса; уменьшение микроглиальной активации; уменьшенная количества амилоидных бляшек | Мыши APP / PSEN1 | [101,102] |
| Эксендин-4 | Снижение ингибирующего фосфорилирования Ser312IRS1, Ser66IRS1 INK, восстановление активация фосфорилирование Tyr465 IRS1
| Культура клеток гиппокампа крыс | [69] |
| Эксендин-4 | Улучшение пространственной памяти в водном лабиринте Морриса; снижение образования амилоида | Мыши APP / PSEN1 | [69] |
| Эксендин-4 - Лираглутид4 | Снижение фосфорилирования eIF2 | Культура клеток гиппокампа крысы, мыши APP / PS1, макаки-крабоеды, введение интрацеребровентрикулярно с Аß -олигомером | [94] |
| ГПП-1 Эксендин-4 | Снижение эксайтотоксичности нейронов | Культура клеток гиппокампа крысы, вводили в базальное ядро с иботеновой кислотой | [103] |
| Розиглитазон | Устранение дефицита памяти в тесте распознавания объектов и при прохождении водного лабиринта Морриса; Снижение уровней Аß -белков | Трансгенные мыши с БА линии J20 Реклама | [104] |
Таблица 2. Репрезентативные доклинические и клинические исследования эффективности противодиабетических препаратов для лечения различных аспектов патологии болезни Альцгеймера
Выводы
Взаимосвязь между СД 3 типа и БА основана на общности биохимических процессов, затрагивающих клиренс Аß-белков и обмен глюкозы в головном мозге.
Кроме того, деградация белка IRS, участвующего в передаче сигналов инсулина, имеет связь с накоплением Аß-белков и формированием бляшек в нервной системе.
Повышение осведомленности о существовании патологического процесса, обозначаемого в данный момент как СД 3 типа, может помочь в оптимизации лечения и профилактики нарушения обмена глюкозы и нейродегенеративных заболеваний мозга.
В настоящее время не существует методов лечения с доказанной эффективностью для борьбы с когнитивными нарушениями при БА, поэтому идентификация БА как расстройства, связанного с передачей сигналов инсулина может иметь важное значение в лечении данного заболевания.
Литература
1. Saeedi, P.; Petersohn, I.; Salpea, P.; Malanda, B.; Karuranga, S.; Unwin, N.; Colagiuri, S.; Guariguata, L.; Motala, A.A.; Ogurtsova, K.; et al. Global and regional diabetes prevalence estimates for 2019 and projections for 2030 and 2045: Results from the International Diabetes Federation Diabetes Atlas, 9th edition. Diabetes Res. Clin. Pract. 2019, 157, 107843.
2. Ngoc, N.B.; Lin, Z.L.; Ahmed, W. Diabetes: What Challenges Lie Ahead for Vietnam? Ann. Glob. Health 2020, 86, 1.
3. Mathers, C.D.; Loncar, D. Projections of global mortality and burden of disease from 2002 to 2030. PLoS Med. 2006, 3, e442.
4. Nguyen, T.T.; Giau, V.V.; Vo, T.K. Current advances in transdermal delivery of drugs for Alzheimer’s disease. Indian J. Pharmacol. 2017, 49, 145–154.
5. Bedse, G.; di Domenico, F.; Serviddio, G.; Cassano, T. Aberrant insulin signaling in Alzheimer’s disease: Current knowledge. Front. Neurosci. 2015, 9, 204.
6. Duarte, J.M.N. Metabolic Alterations Associated to Brain Dysfunction in Diabetes. Aging Dis. 2015, 6, 304–321.
7. Nguyen, T.T.; Ta, Q.T.H.; Nguyen, T.T.D.; Le, T.T.; Vo, V.G. Role of Insulin Resistance in the Alzheimer’s Disease Progression. Neurochem. Res. 2020.
8. Caberlotto, L.; Nguyen, T.P.; Lauria, M.; Priami, C.; Rimondini, R.; Maioli, S.; Cedazo-Minguez, A.; Sita, G.; Morroni, F.; Corsi, M.; et al. Cross-disease analysis of Alzheimer’s disease and type-2 Diabetes highlights the role of autophagy in the pathophysiology of two highly comorbid diseases. Sci. Rep. 2019, 9, 3965.
9. Correia, S.C.; Santos, R.X.; Carvalho, C.; Cardoso, S.; Candeias, E.; Santos, M.S.; Oliveira, C.R.; Moreira, P.I. Insulin signaling, glucose metabolism and mitochondria: Major players in Alzheimer’s disease and diabetes interrelation. Brain Res. 2012, 1441, 64–78.
10. Baker, L.D.; Cross, D.J.; Minoshima, S.; Belongia, D.; Watson, G.S.; Craft, S. Insulin resistance and Alzheimer-like reductions in regional cerebral glucose metabolism for cognitively normal adults with prediabetes or early type 2 diabetes. Arch. Neurol. 2011, 68, 51–57.
11. Ferreira, L.S.S.; Fernandes, C.S.; Vieira, M.N.N.; de Felice, F.G. Insulin Resistance in Alzheimer’s Disease. Front. Neurosci. 2018, 12, 830.
12. Rorbach-Dolata, A.; Piwowar, A. Neurometabolic Evidence Supporting the Hypothesis of Increased Incidence of Type 3 Diabetes Mellitus in the 21st Century. Biomed. Res. Int. 2019, 2019, 8.
13. Ormazabal, V.; Nair, S.; Elfeky, O.; Aguayo, C.; Salomon, C.; Zuñiga, F.A. Association between insulin resistance and the development of cardiovascular disease. Cardiovasc. Diabetol. 2018, 17, 122.
14. Weinstein, G.; Davis-Plourde, K.L.; Conner, S.; Himali, J.J.; Beiser, A.S.; Lee, A.; Rawlings, A.M.; Sedaghat, S.; Ding, J.; Moshier, E.; et al. Association of metformin, sulfonylurea and insulin use with brain structure and function and risk of dementia and Alzheimer’s disease: Pooled analysis from 5 cohorts. PLoS ONE 2019, 14, e0212293.
15. Hubbard, S.R. The insulin receptor: Both a prototypical and atypical receptor tyrosine kinase. Cold Spring Harb. Perspect. Biol. 2013, 5, a008946.
16. Hubbard, S.R. Crystal structure of the activated insulin receptor tyrosine kinase in complex with peptide substrate and ATP analog. Embo J. 1997, 16, 5572–5581.
17. Bosco, D.; Fava, A.; Plastino, M.; Montalcini, T.; Pujia, A. Possible implications of insulin resistance and glucose metabolism in Alzheimer’s disease pathogenesis. J. Cell. Mol. Med. 2011, 15, 1807–1821.
18. Chiu, S.L.; Chen, C.M.; Cline, H.T. Insulin receptor signaling regulates synapse number, dendritic plasticity, and circuit function in vivo. Neuron 2008, 58, 708–719.
19. Lee, C.C.; Huang, C.C.; Hsu, K.S. Insulin promotes dendritic spine and synapse formation by the PI3K/Akt/mTOR and Rac1 signaling pathways. Neuropharmacology 2011, 61, 867–879.
20. Lee, S.-H.; Zabolotny, J.M.; Huang, H.; Lee, H.; Kim, Y.-B. Insulin in the nervous system and the mind: Functions in metabolism, memory, and mood. Mol. Metab. 2016, 5, 589–601.
21. Peineau, S.; Taghibiglou, C.; Bradley, C.; Wong, T.P.; Liu, L.; Lu, J.; Lo, E.; Wu, D.; Saule, E.; Bouschet, T.; et al. LTP inhibits LTD in the hippocampus via regulation of GSK3beta. Neuron 2007, 53, 703–717.
22. Kim, S.J.; Han, Y. Insulin inhibits AMPA-induced neuronal damage via stimulation of protein kinase B (Akt). J. Neural Transm. (Vienna, Austria: 1996) 2005, 112, 179–191.
23. Tomita, T. Apoptosis in pancreatic β-islet cells in Type 2 diabetes. Bosn. J. Basic. Med. Sci. 2016, 16, 162–179.
24. Havrankova, J.; Schmechel, D.; Roth, J.; Brownstein, M. Identification of insulin in rat brain. Proc. Natl. Acad. Sci. USA 1978, 75, 5737–5741.
25. Dorn, A.; Bernstein, H.G.; Rinne, A.; Ziegler, M.; Hahn, H.J.; Ansorge, S. Insulin- and glucagonlike peptides in the brain. Anat. Rec. 1983, 207, 69–77.
26. Young, W.S., 3rd. Periventricular hypothalamic cells in the rat brain contain insulin mRNA. Neuropeptides 1986, 8, 93–97.
27. Gerozissis, K. Brain insulin: Regulation, mechanisms of action and functions. Cell. Mol. Neurobiol. 2003, 23, 1–25.
28. Clarke, D.W.; Mudd, L.; Boyd, F.T., Jr.; Fields, M.; Raizada, M.K. Insulin is released from rat brain neuronal cells in culture. J. Neurochem. 1986, 47, 831–836.
29. Pomytkin, I.; Costa-Nunes, J.P.; Kasatkin, V.; Veniaminova, E.; Demchenko, A.; Lyundup, A.; Lesch, K.-P.; Ponomarev, E.D.; Strekalova, T. Insulin receptor in the brain: Mechanisms of activation and the role in the CNS pathology and treatment. CNS Neurosci. Ther. 2018, 24, 763–774.
30. Hancock, M.L.; Meyer, R.C.; Mistry, M.; Khetani, R.S.; Wagschal, A.; Shin, T.; Sui, S.J.H.; Näär, A.M.; Flanagan, J.G. Insulin Receptor Associates with Promoters Genome-wide and Regulates Gene Expression. Cell 2019, 177, 722–736.e22.
31. Frolich, L.; Blum-Degen, D.; Bernstein, H.G.; Engelsberger, S.; Humrich, J.; Laufer, S.; Muschner, D.; Thalheimer, A.; Turk, A.; Hoyer, S.; et al. Brain insulin and insulin receptors in aging and sporadic Alzheimer’s disease. J. Neural Transm. (Vienna, Austria: 1996) 1998, 105, 423–438.
32. Herrup, K.; Yang, Y. Cell cycle regulation in the postmitotic neuron: Oxymoron or new biology? Nat. Rev. Neurosci. 2007, 8, 368–378.
33. Gubellini, P.; Picconi, B.; di Filippo, M.; Calabresi, P. Downstream mechanisms triggered by mitochondrial dysfunction in the basal ganglia: From experimental models to neurodegenerative diseases. Biochim. Biophys. Acta 2010, 1802, 151–161.
34. Apelt, J.; Mehlhorn, G.; Schliebs, R. Insulin-sensitive GLUT4 glucose transporters are colocalized with GLUT3-expressing cells and demonstrate a chemically distinct neuron-specific localization in rat brain. J. Neurosci. Res. 1999, 57, 693–705.
35. Chen, Z.; Zhong, C. Decoding Alzheimer’s disease from perturbed cerebral glucose metabolism: Implications for diagnostic and therapeutic strategies. Prog. Neurobiol. 2013, 108, 21–43.
36. Liu, Y.; Liu, F.; Iqbal, K.; Grundke-Iqbal, I.; Gong, C.-X. Decreased glucose transporters correlate to abnormal hyperphosphorylation of tau in Alzheimer disease. FEBS Lett. 2008, 582, 359–364.
37. Li, L.; Holscher, C. Common pathological processes in Alzheimer disease and type 2 diabetes: A review. Brain Res. Rev. 2007, 56, 384–402.
38. Goldstein, B.J. Insulin resistance as the core defect in type 2 diabetes mellitus. Am. J. Cardiol. 2002, 90, 3g–10g.
39. Mielke, J.G.; Taghibiglou, C.; Liu, L.; Zhang, Y.; Jia, Z.; Adeli, K.; Wang, Y.T. A biochemical and functional characterization of diet-induced brain insulin resistance. J. Neurochem. 2005, 93, 1568–1578.
40. Hardigan, T.; Ward, R.; Ergul, A. Cerebrovascular complications of diabetes: Focus on cognitive dysfunction. Clin. Sci. (Lond.) 2016, 130, 1807–1822.
41. Hoyer, S. The brain insulin signal transduction system and sporadic (type II) Alzheimer disease: An update. J. Neural Transm. (Vienna, Austria: 1996) 2002, 109, 341–360.
42. Farris, W.; Mansourian, S.; Chang, Y.; Lindsley, L.; Eckman, E.A.; Frosch, M.P.; Eckman, C.B.; Tanzi, R.E.; Selkoe, D.J.; Guénette, S. Insulin-degrading enzyme regulates the levels of insulin, amyloid β-protein, and the β-amyloid precursor protein intracellular domain in vivo. Proc. Natl. Acad. Sci. USA 2003, 100, 4162–4167.
43. de la Monte, S.M. Brain insulin resistance and deficiency as therapeutic targets in Alzheimer’s disease. Curr. Alzheimer Res. 2012, 9, 35–66.
44. Ling, X.; Martins, R.N.; Racchi, M.; Craft, S.; Helmerhorst, E. Amyloid beta antagonizes insulin promoted secretion of the amyloid beta protein precursor. J. Alzheimer’s Dis. Jad. 2002, 4, 369–374.
45. Zheng, W.H.; Kar, S.; Quirion, R. Insulin-like growth factor-1-induced phosphorylation of the forkhead family transcription factor FKHRL1 is mediated by Akt kinase in PC12 cells. J. Biol. Chem. 2000, 275, 39152–39158.
46. Gasparini, L.; Gouras, G.K.; Wang, R.; Gross, R.S.; Beal, M.F.; Greengard, P.; Xu, H. Stimulation of β-Amyloid Precursor Protein Trafficking by Insulin Reduces Intraneuronal β-Amyloid and Requires Mitogen-Activated Protein Kinase Signaling. J. Neurosci. 2001, 21, 2561–2570.
47. Delikkaya, B.; Moriel, N.; Tong, M.; Gallucci, G.; de la Monte, S.M. Altered expression of insulin-degrading enzyme and regulator of calcineurin in the rat intracerebral streptozotocin model and human apolipoprotein E-ε4–associated Alzheimer’s disease, Alzheimer’s & Dementia: Diagnosis. Assess. Dis. Monit. 2019, 11, 392–404.
48. Mittal, K.; Mani, R.J.; Katare, D.P. Type 3 Diabetes: Cross Talk between Differentially Regulated Proteins of Type 2 Diabetes Mellitus and Alzheimer’s Disease. Sci. Rep. 2016, 6, 25589.
49. Kroner, Z. The relationship between Alzheimer’s disease and diabetes: Type 3 diabetes? Altern. Med. Rev. A J. Clin. Ther. 2009, 14, 373–379.
50. Talbot, K.; Wang, H.Y.; Kazi, H.; Han, L.Y.; Bakshi, K.P.; Stucky, A.; Fuino, R.L.; Kawaguchi, K.R.; Samoyedny, A.J.; Wilson, R.S.; et al. Demonstrated brain insulin resistance in Alzheimer’s disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. J. Clin. Investig. 2012, 122, 1316–1338
51. de la Monte, S.M. Type 3 diabetes is sporadic Alzheimer’s disease: Mini-review. Eur. Neuropsychopharmacol. J. Eur. Coll. Neuropsychopharmacol. 2014, 24, 1954–1960.
52. Kivipelto, M.; Ngandu, T.; Fratiglioni, L.; Viitanen, M.; Kareholt, I.; Winblad, B.; Helkala, E.L.; Tuomilehto, J.; Soininen, H.; Nissinen, A. Obesity and vascular risk factors at midlife and the risk of dementia and Alzheimer disease. Arch. Neurol. 2005, 62, 1556–1560.
53. Razay, G.; Vreugdenhil, A.; Wilcock, G. Obesity, abdominal obesity and Alzheimer disease. Dement. Geriatr. Cogn. Disord. 2006, 22, 173–176.
54. Kullmann, S.; Heni, M.; Hallschmid, M.; Fritsche, A.; Preissl, H.; Haring, H.U. Brain Insulin Resistance at the Crossroads of Metabolic and Cognitive Disorders in Humans. Physiol. Rev. 2016, 96, 1169–1209
55. Gabbouj, S.; Ryhänen, S.; Marttinen, M.; Wittrahm, R.; Takalo, M.; Kemppainen, S.; Martiskainen, H.; Tanila, H.; Haapasalo, A.; Hiltunen, M.; et al. Altered Insulin Signaling in Alzheimer’s Disease Brain—Special Emphasis on PI3K-Akt Pathway. Front. Neurosci. 2019, 13, 629.
56. Lillioja, S.; Mott, D.M.; Spraul, M.; Ferraro, R.; Foley, J.E.; Ravussin, E.; Knowler, W.C.; Bennett, P.H.; Bogardus, C. Insulin resistance and insulin secretory dysfunction as precursors of non-insulin-dependent diabetes mellitus. Prospective studies of Pima Indians. N. Engl. J. Med. 1993, 329, 1988–1992
57. Li, J.; Bai, L.; Wei, F.; Zhao, J.; Wang, D.; Xiao, Y.; Yan, W.; Wei, J. Therapeutic Mechanisms of Herbal Medicines Against Insulin Resistance: A Review. Front. Pharmacol. 2019, 10, 661
58. Kapogiannis, D.; Boxer, A.; Schwartz, J.B.; Abner, E.L.; Biragyn, A.; Masharani, U.; Frassetto, L.; Petersen, R.C.; Miller, B.L.; Goetzl, E.J. Dysfunctionally phosphorylated type 1 insulin receptor substrate in neural-derived blood exosomes of preclinical Alzheimer’s disease. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2015, 29, 589–596.
59. Fontaine, J.F.; Barbosa-Silva, A.; Schaefer, M.; Huska, M.R.; Muro, E.M.; Andrade-Navarro, M.A. MedlineRanker: Flexible ranking of biomedical literature. Nucleic Acids Res. 2009, 37, W141–W146.
60. Wang, G. Raison d’être of insulin resistance: The adjustable threshold hypothesis. J. R. Soc. Interface 2014, 11, 20140892
61. Nisr, R.B.; Affourtit, C. Insulin acutely improves mitochondrial function of rat and human skeletal muscle by increasing coupling efficiency of oxidative phosphorylation. Biochim. Biophys. Acta 2014, 1837, 270–276.
62. Sivitz, W.I.; Yorek, M.A. Mitochondrial dysfunction in diabetes: From molecular mechanisms to functional significance and therapeutic opportunities. Antioxid. Redox Signal. 2010, 12, 537–577
63. Bucht, G.; Adolfsson, R.; Lithner, F.; Winblad, B. Changes in blood glucose and insulin secretion in patients with senile dementia of Alzheimer type. Acta Med. Scand. 1983, 213, 387–392
64. Matioli, M.N.P.S.; Nitrini, R. Mechanisms linking brain insulin resistance to Alzheimer’s disease. Dement. Neuropsychol. 2015, 9, 96–102
65. Ma, Q.L.; Yang, F.; Rosario, E.R.; Ubeda, O.J.; Beech, W.; Gant, D.J.; Chen, P.P.; Hudspeth, B.; Chen, C.; Zhao, Y.; et al. Beta-amyloid oligomers induce phosphorylation of tau and inactivation of insulin receptor substrate via c-Jun N-terminal kinase signaling: Suppression by omega-3 fatty acids and curcumin. J. Neurosci. Off. J. Soc. Neurosci. 2009, 29, 9078–9089.
66. Velazquez, R.; Tran, A.; Ishimwe, E.; Denner, L.; Dave, N.; Oddo, S.; Dineley, K.T. Central insulin dysregulation and energy dyshomeostasis in two mouse models of Alzheimer’s disease. Neurobiol. Aging 2017, 58, 1–13
67. Ruiz, H.H.; Chi, T.; Shin, A.C.; Lindtner, C.; Hsieh, W.; Ehrlich, M.; Gandy, S.; Buettner, C. Increased susceptibility to metabolic dysregulation in a mouse model of Alzheimer’s disease is associated with impaired hypothalamic insulin signaling and elevated BCAA levels. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2016, 12, 851–861.
68. Long-Smith, C.M.; Manning, S.; McClean, P.L.; Coakley, M.F.; O’Halloran, D.J.; Holscher, C.; O’Neill, C. The diabetes drug liraglutide ameliorates aberrant insulin receptor localisation and signalling in parallel with decreasing both amyloid-beta plaque and glial pathology in a mouse model of Alzheimer’s disease. Neuromol. Med. 2013, 15, 102–114.
69. Bomfim, T.R.; Forny-Germano, L.; Sathler, L.B.; Brito-Moreira, J.; Houzel, J.C.; Decker, H.; Silverman, M.A.; Kazi, H.; Melo, H.M.; McClean, P.L.; et al. An anti-diabetes agent protects the mouse brain from defective insulin signaling caused by Alzheimer’s disease- associated Abeta oligomers. J. Clin. Investig. 2012, 122, 1339–1353.
70. Clarke, J.R.; Lyra, E.S.N.M.; Figueiredo, C.P.; Frozza, R.L.; Ledo, J.H.; Beckman, D.; Katashima, C.K.; Razolli, D.; Carvalho, B.M.; Frazao, R.; et al. Alzheimer-associated Abeta oligomers impact the central nervous system to induce peripheral metabolic deregulation. Embo Mol. Med. 2015, 7, 190–210.
71. Tanokashira, D.; Fukuokaya, W.; Taguchi, A. Involvement of insulin receptor substrates in cognitive impairment and Alzheimer’s disease. Neural Regen. Res. 2019, 14, 1330–1334.
72. Freude, S.; Hettich, M.M.; Schumann, C.; Stohr, O.; Koch, L.; Kohler, C.; Udelhoven, M.; Leeser, U.; Muller, M.; Kubota, N.; et al. Neuronal IGF-1 resistance reduces Abeta accumulation and protects against premature death in a model of Alzheimer’s disease. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2009, 23, 3315–3324.
73. Taguchi, A.; Wartschow, L.M.; White, M.F. Brain IRS2 signaling coordinates life span and nutrient homeostasis. Science (N. Y.) 2007, 317, 369–372.
74. Killick, R.; Scales, G.; Leroy, K.; Causevic, M.; Hooper, C.; Irvine, E.E.; Choudhury, A.I.; Drinkwater, L.; Kerr, F.; Al-Qassab, H.; et al. Deletion of Irs2 reduces amyloid deposition and rescues behavioural deficits in APP transgenic mice. Biochem. Biophys. Res. Commun. 2009, 386, 257–262
75. Kandimalla, R.; Thirumala, V.; Reddy, P.H. Is Alzheimer’s disease a Type 3 Diabetes? A critical appraisal. Biochim. Biophys. Acta. Mol. Basis Dis. 2017, 1863, 1078–1089.
76. Nguyen, N.H.; Pham, Q.T.; Luong, T.N.H.; Le, H.K.; Vo, V.G. Potential Antidiabetic Activity of Extracts and Isolated Compound from Adenosma bracteosum (Bonati). Biomolecules 2020, 10, 201.
77. Giau, V.V.; Wu, S.Y.; Jamerlan, A.; An, S.S.A.; Kim, S.; Hulme, J. Gut Microbiota and Their Neuroinflammatory Implications in Alzheimer’s Disease. Nutrients 2018, 10, 1765.
78. de Matos, A.M.; de Macedo, M.P.; Rauter, A.P. Bridging Type 2 Diabetes and Alzheimer’s Disease: Assembling the Puzzle Pieces in the Quest for the Molecules With Therapeutic and Preventive Potential. Med. Res. Rev. 2018, 38, 261–324.
79. Bagyinszky, E.; Giau, V.V.; Shim, K.; Suk, K.; An, S.S.A.; Kim, S. Role of inflammatory molecules in the Alzheimer’s disease progression and diagnosis. J. Neurol. Sci. 2017, 376, 242–254
80. van Giau, V.; An, S.S.A.; Hulme, J.P. Mitochondrial therapeutic interventions in Alzheimer’s disease. J. Neurol. Sci. 2018, 395, 62–70
81. Ayaz, M.; Sadiq, A.; Junaid, M.; Ullah, F.; Ovais, M.; Ullah, I.; Ahmed, J.; Shahid, M. Flavonoids as Prospective Neuroprotectants and Their Therapeutic Propensity in Aging Associated Neurological Disorders. Front. Aging Neurosci. 2019, 11.
82. Canhada, S.; Castro, K.; Perry, I.S.; Luft, V.C. Omega-3 fatty acids’ supplementation in Alzheimer’s disease: A systematic review. Nutr. Neurosci. 2018, 21, 529–538
83. Ajith, T.A. A Recent Update on the Effects of Omega-3 Fatty Acids in Alzheimer’s Disease. Curr. Clin. Pharmacol. 2018, 13, 252–260.
84. Broom, G.M.; Shaw, I.C.; Rucklidge, J.J. The ketogenic diet as a potential treatment and prevention strategy for Alzheimer’s disease. Nutrition (Burbank, Los Angeles County, Calif.) 2019, 60, 118–121
85. Giau, V.V.; Bagyinszky, E.; An, S.S.A.; Kim, S.Y. Role of apolipoprotein E in neurodegenerative diseases. Neuropsychiatr. Dis. Treat. 2015, 11, 1723–1737
86. Stoykovich, S.; Gibas, K. APOE ε4, the door to insulin-resistant dyslipidemia and brain fog? A case study. Alzheimers Dement. (Amst) 2019, 11, 264–269
87. Zhao, N.; Liu, C.C.; van Ingelgom, A.J.; Martens, Y.A.; Linares, C.; Knight, J.A.; Painter, M.M.; Sullivan, P.M.; Bu, G. Apolipoprotein E4 Impairs Neuronal Insulin Signaling by Trapping Insulin Receptor in the Endosomes. Neuron 2017, 96, 115–129.e5.
88. Frederiksen, K.S.; Gjerum, L.; Waldemar, G.; Hasselbalch, S.G. Effects of Physical Exercise on Alzheimer’s Disease Biomarkers: A Systematic Review of Intervention Studies. J. Alzheimer’s Dis. 2018, 61, 359–372.
89. Li, C.; Liu, W.; Li, X.; Zhang, Z.; Qi, H.; Liu, S.; Yan, N.; Xing, Y.; Holscher, C.; Wang, Z. The novel GLP-1/GIP analogue DA5-CH reduces tau phosphorylation and normalizes theta rhythm in the icv. STZ rat model of AD. Brain Behav. 2020, 10, e01505
90. Wang, L.; Zhang, R.; Hou, X.; Wang, C.; Guo, S.; Ning, N.; Sun, C.; Yuan, Y.; Li, L.; Hölscher, C.; et al. DA-JC1 improves learning and memory by antagonizing Aβ31–35-induced circadian rhythm disorder. Mol. Brain 2019, 12, 14
91. Cao, Y.; Holscher, C.; Hu, M.M.; Wang, T.; Zhao, F.; Bai, Y.; Zhang, J.; Wu, M.N.; Qi, J.S. DA5-CH, a novel GLP-1/GIP dual agonist, effectively ameliorates the cognitive impairments and pathology in the APP/PS1 mouse model of Alzheimer’s disease. Eur. J. Pharmacol. 2018, 827, 215–226
92. Panagaki, T.; Gengler, S.; Holscher, C. The Novel DA-CH3 Dual Incretin Restores Endoplasmic Reticulum Stress and Autophagy Impairments to Attenuate Alzheimer-Like Pathology and Cognitive Decrements in the APPSWE/PS1DeltaE9 Mouse Model. J. Alzheimer’s Dis. 2018, 66, 195–218
93. de Felice, F.G.; Vieira, M.N.; Bomfim, T.R.; Decker, H.; Velasco, P.T.; Lambert, M.P.; Viola, K.L.; Zhao, W.Q.; Ferreira, S.T.; Klein, W.L. Protection of synapses against Alzheimer’s-linked toxins: Insulin signaling prevents the pathogenic binding of Abeta oligomers. Proc. Natl. Acad. Sci. USA 2009, 106, 1971–1976.
94. Lourenco, M.V.; Clarke, J.R.; Frozza, R.L.; Bomfim, T.R.; Forny-Germano, L.; Batista, A.F.; Sathler, L.B.; Brito-Moreira, J.; Amaral, O.B.; Silva, C.A.; et al. TNF-alpha mediates PKR-dependent memory impairment and brain IRS-1 inhibition induced by Alzheimer’s beta-amyloid oligomers in mice and monkeys. Cell Metab. 2013, 18, 831–843.
95. Craft, S.; Asthana, S.; Cook, D.G.; Baker, L.D.; Cherrier, M.; Purganan, K.; Wait, C.; Petrova, A.; Latendresse, S.; Watson, G.S.; et al. Insulin dose-response effects on memory and plasma amyloid precursor protein in Alzheimer’s disease: Interactions with apolipoprotein E genotype. Psychoneuroendocrinology 2003, 28, 809–822.
96. Reger, M.A.; Watson, G.S.; Green, P.S.; Baker, L.D.; Cholerton, B.; Fishel, M.A.; Plymate, S.R.; Cherrier, M.M.; Schellenberg, G.D.; Frey, W.H., 2nd; et al. Intranasal insulin administration dose-dependently modulates verbal memory and plasma amyloid-beta in memory-impaired older adults. J. Alzheimer’s Dis. 2008, 13, 323–331.
97. Reger, M.A.; Watson, G.S.; Frey, W.H., 2nd; Baker, L.D.; Cholerton, B.; Keeling, M.L.; Belongia, D.A.; Fishel, M.A.; Plymate, S.R.; Schellenberg, G.D.; et al. Effects of intranasal insulin on cognition in memory-impaired older adults: Modulation by APOE genotype. Neurobiol. Aging 2006, 27, 451–458.
98. Reger, M.A.; Watson, G.S.; Green, P.S.; Wilkinson, C.W.; Baker, L.D.; Cholerton, B.; Fishel, M.A.; Plymate, S.R.; Breitner, J.C.; DeGroodt, W.; et al. Intranasal insulin improves cognition and modulates beta-amyloid in early AD. Neurology 2008, 70, 440–448
99. Benedict, C.; Kern, W.; Schultes, B.; Born, J.; Hallschmid, M. Differential sensitivity of men and women to anorexigenic and memory-improving effects of intranasal insulin. J. Clin. Endocrinol. Metab. 2008, 93, 1339–1344
100. Batista, A.F.; Forny-Germano, L.; Clarke, J.R.; Lyra, E.S.N.M.; Brito-Moreira, J.; Boehnke, S.E.; Winterborn, A.; Coe, B.C.; Lablans, A.; Vital, J.F.; et al. The diabetes drug liraglutide reverses cognitive impairment in mice and attenuates insulin receptor and synaptic pathology in a non-human primate model of Alzheimer’s disease. J. Pathol. 2018, 245, 85–100
101. McClean, P.L.; Parthsarathy, V.; Faivre, E.; Holscher, C. The diabetes drug liraglutide prevents degenerative processes in a mouse model of Alzheimer’s disease. J. Neurosci. Off. J. Soc. Neurosci. 2011, 31, 6587–6594
102. McClean, P.L.; Holscher, C. Liraglutide can reverse memory impairment, synaptic loss and reduce plaque load in aged APP/PS1 mice, a model of Alzheimer’s disease. Neuropharmacology 2014, 76 Pt A, 57–67
103. Perry, T.; Haughey, N.J.; Mattson, M.P.; Egan, J.M.; Greig, N.H. Protection and reversal of excitotoxic neuronal damage by glucagon-like peptide-1 and exendin-4. J. Pharmacol. Exp. Ther. 2002, 302, 881–888
104. Escribano, L.; Simon, A.M.; Gimeno, E.; Cuadrado-Tejedor, M.; de Maturana, R.L.; Garcia-Osta, A.; Ricobaraza, A.; Perez-Mediavilla, A.; del Rio, J.; Frechilla, D. Rosiglitazone rescues memory impairment in Alzheimer’s transgenic mice: Mechanisms involving a reduced amyloid and tau pathology. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 2010, 35, 1593–1604