Начиная с 60–70-х гг. прошлого века случаи стремительно развивающейся почечной недостаточности у больных, исходно не имевших патологии почек, традиционно расценивались как гломерулонефрит с логически вытекающей из этого диагноза попыткой остановить прогрессирование заболевания применением интенсивной иммуносупрессивной терапии. Однако в повседневной практике врача нередко встречаются пациенты, течение болезни у которых напоминает форму быстропрогрессирующего гломерулонефрита (БПГН), хотя по своей сути эта болезнь гломерулонефритом не является. Их отличают некоторые особенности развития заболевания, чаще всего ранняя и трудноконтролируемая артериальная гипертензия (АГ), сочетающаяся с неврологическими нарушениями (головные боли, транзиторные ишемические атаки или острое нарушение мозгового кровообращения в молодом возрасте), рецидивирующими осложнениями беременности у женщин, тромбозами, в том числе необычной локализации. Наследственный анамнез этих больных нередко отягощен по сердечно-сосудистым катастрофам и тромбозам. Другими особенностями, свойственными этим пациентам, можно считать гиперкоагуляционные изменения при исследовании системы гемостаза, иногда с признаками активации внутрисосудистого свертывания крови.
Все это даже при казалось бы типичной клинической картине другого заболевания, например нефрита, позволяет заподозрить у таких больных сосудистую патологию почек, в первую очередь тромботическую микроангиопатию (ТМА), даже при отсутствии микроангиопатической гемолитической анемии и тромбоцитопении.
Тромботическая микроангиопатия (ТМА) представляет собой особый тип поражения мелких внутриорганных сосудов, преимущественно почечных. Характеризуется сочетанием острых тромбозов, отека эндотелиальных клеток с отслойкой от базальной мембраны, утолщения сосудистой стенки с хроническими сосудистыми изменениями, включающими фиброзную гиперплазию интимы, артерио- и артериолосклероз и организующиеся тромбы с реканализацией или без нее, что в конечном итоге приводит к фиброзной окклюзии пораженных сосудов и может вызвать ишемическую атрофию коры почек вследствие нарушений перфузии. Учитывая однотипность гистологических изменений почек при любых заболеваниях, в основе которых лежит ТМА, в большинстве случаев биопсия почки не позволяет дифференцировать формы ТМА друг от друга, несмотря на различия механизмов повреждения, приводящих к микроангиопатическим тромбозам. Однако независимо от того, развивается ли ТМА первично или вторично, центральным звеном патогенеза является повреждение сосудистого эндотелия в органах-мишенях, главным образом в почках. При этом клинические проявления в виде быстро нарастающей почечной недостаточности, тяжелой АГ, выраженной микрогематурии и даже массивной протеинурии не отличаются от таковых при быстропрогрессирующем гломерулонефрите (БПГН) [1].
До недавнего времени были известны практически лишь две формы ТМА �� гемолитико-уремический синдром (ГУС) и тромботическая тромбоцитопеническая пурпура (ТТП), не всегда отличимые друг от друга у взрослых пациентов. В 2006 году Besbas и соавт. предложили новую классификацию, в которую вошли все известные на сегодняшний день состояния, ассоциированные с развитием ТМА [2]. В настоящее время выделяют ТМА, ассоциированную с инфекцией, с генетическими или приобретенными нарушениями регуляторных белков системы комплемента (атипичный ГУС) и с дефицитом протеиназы, расщепляющей фактор Виллебранда (ADAMTS-13) (ТТП). ТМА, индуцированная инфекцией, — это, в первую очередь, типичный постдиарейный ГУС, вызванный веротоксин-продуцирующей Еscherichia coli (VTEC) или Shigella dysenteriae I типа, хотя к этой группе можно отнести также ТМА, ассоциированную с инфекцией Citrobacter и Streptococcus pneumoniae. Наряду с упомянутыми формами выделены формы, связанные с ВИЧ-инфекцией, злокачественными опухолями, их химио- и радиотерапией, с лечением ингибиторами кальцийнейрина и трансплантацией органов, беременностью и родами (преэклампсия-эклампсия, HELLP-синдром), злокачественной артериальной гипертензией (АГ), с приемом оральных контрацептивов, развивающиеся при аутоиммунных заболеваниях (системной красной волчанке, склеродермии, антифосфолипидном синдроме). Эти формы, по-видимому, можно считать вторичными, однако пока их патогенез остается неуточненным.
В последние годы получены данные, свидетельствующие о возможности тромботического поражения сосудистого русла почек при наиболее частых генетических формах тромбофилии — лейденской мутации V фактора свертывания крови, мутациях генов протромбина, ингибитора активатора плазминогена I типа (PAI-I) и метилентетрагидрофолатредуктазы (MTHFR) [3, 4]. Так, Т. Raife и соавт. обнаружили, что у пациентов с клиническими и морфологическими признаками ТМА частота выявления мутации фактора V Leiden была достоверно выше по сравнению с контрольной группой. Кроме того, была выявлена тесная связь развития классических форм ТМА — ГУС и ТТП с носительством генотипа Т/Т гена MTHFR C 677 T и генотипа G/G гена, активируемого тромбином ингибитора фибринoлиза (TAFI G505A), в исследованиях Sucker и соавт. [5].
В наших недавних исследованиях было установлено, что ТМА у пациентов с генетическими тромбофилиями, аналогично нефропатии, ассоциированной с антифосфолипидным синдромом (АФС), может развиваться либо как единственная форма поражения почек, либо сочетаться с уже существующей нефропатией, чаще всего хроническим гломерулонефритом (ХГН) [6]. Однако возможности развития этой патологии мало известны практическим врачам. Проиллюстрировать вышесказанное можно следующими примерами из нашей клинической практики.
Больная Д., 34 лет, обратилась в клинику для уточнения диагноза с жалобами на повышение артериального давления (АД) до 160/90–100 мм рт. ст., плохо уступающее приему антигипертензивных средств (ингибиторы ангиотензинпревращающего фермента (иАПФ), блокаторы рецепторов ангиотензина II (БРА), блокаторы кальциевых каналов, агонисты I1-имидазолиновых рецепторов — рилменидин), отеки, преимущественно голеней.
Из анамнеза: впервые изменения в анализах мочи выявлены в 2004 г., был диагностирован ХГН, в том же году наступившая беременность была прервана по медицинским показаниям на сроке 17–18 недель (документы не сохранились, известно лишь, что почки при УЗИ были нормальных размеров). Иммуносупрессивная терапия не проводилась. Впоследствии, в связи с персистирующим мочевым синдромом, была проведена биопсия почки, получено 14 клубочков, 4 из которых полностью склерозированы, в 5 клубочках в просвете капсулы массивные синехии между капсулой и капиллярными петлями, в большинстве клубочков значительное утолщение базальных мембран клубочков (БМК) и выраженный склероз мезангия, пролиферация мезангиальных клеток, очаговое утолщение и удвоение БМК. Строма вблизи склерозированных клубочков с очаговым фиброзом, лимфоцитарными и макрофагальными инфильтратами. Канальцевая система в очагах фиброза с очаговой деформацией, очаговым сужением и дилатацией — формирующаяся «щитовидная почка». Иммуногистохимическое исследование не проводилось. На светооптическом уровне изменения расценены как мезангиокапиллярный гломерулонефрит (МКГН). В связи с невысоким (0,5 г/л) уровнем протеинурии и стабильно нормальным уровнем АД активная терапия не проводилась.
В 2008 г. — вторая беременность, протекавшая без изменений в анализах мочи со стабильно нормальным уровнем АД, закончившаяся нормальными родами в срок, сын здоров. Сразу после родоразрешения пациентку стали беспокоить сильные головные боли, по поводу которых она обратилась к врачу лишь в 2010 г.: было зарегистрировано повышение АД до 170/120 мм рт. ст., преходящее повышение уровня креатинина крови без значимой протеинурии. Последовательное назначение иАПФ и БРА к нормализации и даже стабилизации АД не привело, БРА были отменены в марте 2011 из-за гиперкалиемии. Назначены амлодипин и релминидин, фуросемид, но стабилизации АД достичь не удалось.
При осмотре 23.06.12 г. обращали на себя внимание сетчатое ливедо на коже конечностей и боковых поверхностей живота, пастозность голеней. При аускультации тоны сердца приглушены, аритмичные, небольшая тахикардия, частота сердечных сокращений (ЧСС) 89 уд./мин, АД 150/100 мм рт. ст. В анализах: стойко высокий (136 г/л), для регистрируемого уровня клубочковой фильтрации, гемоглобин крови, нормальный уровень (4,09 млн/мкл) эритроцитов с нормохромией, гематокрит — 39,5%, лейкоциты — 7,1 тыс./мкл без изменений в формуле, тромбоциты — 204 тыс./мкл, повышение СОЭ до 37 мм/ч. Протеинурия 2,0 г/л, осадок мочи — без особенностей. По данным коагулограммы: D-димер повышен до 1,750 мг/мл (более 3N), волчаночный антикоагулянт не обнаружен. Титр антител к кардиолипину в пределах нормы.
При УЗИ почки уменьшены в размерах: правая — 7,7×3,4×4,0 см, толщина паренхимы 0,9 см, левая — 7,7×4,9×4,0 см, толщина паренхимы 0,9 см. Кортикомедуллярная дифференциация отсутствует. Дифференциация паренхима/почечный синус умеренно снижена. При ультразвуковой ангиографии — обеднение коркового кровотока в режиме цветового допплеровского картирования (ЦДК).
Особенности течения заболевания (быстрое значительное снижение фильтрационной функции почек, связь с беременностью и родами, скудные изменения в анализах мочи с относительно небольшой протеинурией), в сочетании с признаками активации внутрисосудистого свертывания крови (D-димер более 3N) и морфологической картиной почечного биоптата, на светооптическом уровне соответствующей мезангиокапиллярному гломерулонефриту (МКГН), позволили предполагать наличие у пациентки хронической ТМА как причины почечной недостаточности.
Ультразвуковая допплерография (УЗДГ) почечных сосудов, проведенная для уточнения состояния почечного кровотока, выявила снижение пиковых скоростей кровотока в магистральных почечных и интрапаренхиматозных артериях с обеих сторон, свидетельствующее о выраженной ишемии, что косвенно подтверждало высказанное предположение. Мультиспиральная компьютерная томография (МСКТ) почек для уточнения диагноза не проводилась из-за выраженного снижения клубочковой фильтрации до 24,6 мл/мин.
Причина ТМА оставалась неясной, отсутствие анемии и тромбоцитопении позволили исключить ГУС/ТТП. Было высказано предположение о наследственной тромбофилии, с которой могло быть связано микроангиопатическое поражение почек. Для проверки этого предположения выполнено генетическое исследование, выявившее носительство неблагоприятных генотипов, ассоциированных с повышенным риском тромбообразования (гетерозиготные мутации фактора V — лейденская мутация, коагуляционных факторов XII, XIII, гена бета-цепи фибриногена (FGB), MTHFR, PAI-I), что послужило основанием для диагностики мультигенной тромбофилии.
В связи с этим было принято решение о назначении низкомолекулярных гепаринов (НМГ) (фраксипарин 0,3–0,6 мл/сут) на длительный срок под контролем активированного частичного тромбопластинового времени (АЧТВ), D-димера, тромбоцитов крови, суточной протеинурии, биохимических показателей. Для коррекции АГ оставлены рилменидин 2 мг/сут, блокаторы кальциевых каналов под контролем ЧСС, фуросемид.
В первый же месяц лечения АД стабилизировалось на нормальных цифрах с редкими подъемами до 140/100 мм рт. ст., на фоне монотерапии рилменидином в уменьшенной до 1 мг дозе, исчезли отеки. На третьем месяце терапии больная перенесла гипертонический криз с преходящим ухудшением функции почек. К четвертому месяцу от начала лечения отмечались стойкая нормализация функции почек, стабилизация АД на уровне 120–130/80 мм рт. ст. при приеме низких доз антигипертензивных средств, исчезновение протеинурии без применения иммуносупрессии, иАПФ и БРА у больной с диагнозом «хронический гломерулонефрит в стадии хронической почечной недостаточности (ХПН)» (табл.).
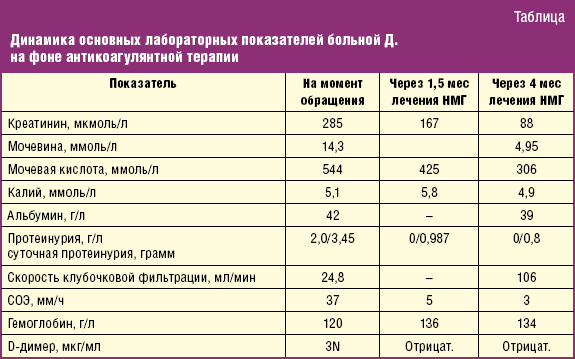
Контрольная УЗДГ почечных сосудов выявила относительное повышение значений пиковых систолических скоростей кровотока в магистральных почечных и интрапаренхиматозных артериях с обеих сторон с нормализацией индексов периферического сопротивления кровотоку как на магистральном, так и на интрапаренхиматозном уровне, что свидетельствует об улучшении микроциркуляции обеих почек в ходе лечения.
Представленное наблюдение с отчетливым положительным эффектом антикоагулянтной терапии, причем не только на уровень азотистых шлаков крови и АД, но и на протеинурию и скорость клубочковой фильтрации вплоть до нормализации их значений, является косвенным свидетельством ишемического повреждения почек тромботического генеза у данной больной. Принимая во внимание несоответствие течения заболевания клинической картине МКГН, активацию внутрисосудистого свертывания крови в отсутствие нефротического синдрома, быстрый ответ на антикоагулянтную терапию, приведшую к полному восстановлению почечной функции, диагноз хронического гломерулонефрита, установленный пациентке на предварительном этапе, представляется неверным. Наиболее обоснован, с нашей точки зрения, диагноз хронической ТМА, несмотря на отсутствие признаков микроангиопатической гемолитической анемии (МАГА) и тромбоцитопении.
В последнее время появились сообщения о развитии ТМА у больных в отсутствии ГУС/ТТП и АФС, протекающей аналогично тому, что мы наблюдали у нашей пациентки. Более того, показано, что в отсутствие тромбоцитопении больные хронической ТМА имеют худший прогноз из-за трудностей диагностики, поскольку диагноз ТМА не включается в круг обсуждаемых заболеваний [7]. Данному диагнозу не противоречит картина мезангиокапиллярного гломерулонефрита, выявленная на светооптическом уровне, поскольку, как известно, целый ряд состояний (болезнь неамилоидного отложения легких цепей, диабетическая нефропатия) наряду с хронической ТМА могут имитировать МКГН. Для дифференциальной диагностики МКГН и перечисленных состояний необходимо проведение иммуногистохимического и электронно-микроскопического исследований, не выполненных в данном случае.
Мы полагаем, что причиной развития ТМА с признаками ренальной дисфункции у представленной больной являются рецидивирующие тромбозы микроциркуляторного русла почек, а имеющуюся у нее мультигенную тромбофилию, по-видимому, следует считать фактором, предрасполагающим к тромбообразованию и его рецидивированию под влиянием классических провоцирующих факторов (беременность и роды). Особенности течения нефропатии, в том числе отсутствие эритроцитурии, изолированный характер протеинурии при морфологической картине МКГН, АГ, резистентная к трем антигипертензивным препаратам, эпизоды рецидивирующего повышения уровня креатинина и, наконец, срок достижения ХПН (6 лет от дебюта болезни) при относительно небольшой протеинурии, заставляли усомниться в диагнозе ХГН и свидетельствовали, скорее, о сосудистой природе заболевания, проявляющейся симптомами ишемии почечной ткани. Несмотря на отсутствие тромбоза капилляров клубочков и мелких внегломерулярных сосудов, имеющиеся гистологические изменения позволяют предполагать наличие хронической ТМА, аналогичной АФС. Отсутствие серологических маркеров АФС побудило нас искать иную причину тромбофилии, которой оказались множественные полиморфизмы в генах свертывания крови. Как было показано в наших предыдущих исследованиях, носительство трех и более мутантных аллелей в генах гемостаза, особенно комбинация замен в генах FGB, PAI-I и MTHFR, что имело место у нашей пациентки, вызывает формирование гиперкоагуляционного состояния с активацией внутрисосудистого свертывания крови в мелких внутрипочечных сосудах [8] и способно, по-видимому, привести к развитию ТМА.
Представленное наблюдение призвано привлечь внимание практических врачей к нефропатиям, протекающим с относительно небольшой протеинурией, ранним повышением уровня креатинина, с довольно высоким темпом прогрессирования почечной недостаточности, напоминающим БПГН. Однако в случаях отсутствия классической клинической картины последнего, генетическое исследование, направленное на верификацию тромбофилии, наряду с биопсией почки может быть использовано как дополнительный диагностический инструмент в диагностике ТМА.
В отличие от первой пациентки, во втором представляемом случае течение болезни сопровождалось яркими системными проявлениями.
Больной 29 лет. Данные о семейном анамнезе отсутствуют. Курит. В возрасте 28 лет (2007 г.) стали беспокоить головные боли, зафиксировано повышение АД до 200/100 мм рт. ст., к врачам не обращался, при повышении АД самостоятельно принимал Энап. В августе 2009 г. после пребывания в жарком климате появился навязчивый сухой кашель, одышка в покое. При обследовании: АД 200/120 мм рт. ст., анемия (гемоглобин 104 г/л) и выраженная азотемия (содержание креатинина в сыворотке крови (Скр) 6,7 мг/дл). При эхокардиографии выявлены расширение полостей левого желудочка и левого предсердия, диффузный гипокинез и умеренная гипертрофия миокарда левого желудочка, фракция выброса 43%, утолщение створок митрального клапана, признаки легочной гипертензии, небольшое количество жидкости в полости перикарда. Диагностирована злокачественная АГ, обсуждался диагноз миокардита. Назначена антигипертензивная терапия (диуретики, бета-адреноблокаторы, иАПФ), в результате которой АД снизилось до 150/80 мм рт. ст. Обследование было продолжено в Городской клинической больнице имени С. П. Боткина (г. Москва), где выявлены: усугубление анемии (гемоглобин 91 г/л), тромбоцитопения (153 тыс./мкл), повышение уровня лактатдегидрогеназы (ЛДГ) до 724 ед/л, минимальный мочевой синдром (протеинурия 0,25 г/л) без изменения мочевого осадка, при сохранном диурезе (1500 мл/сут) отмечено дальнейшее нарастание уровня креатинина (Скр 7,5 мг/дл). При рентгенологическом исследовании отмечалось усиление легочного рисунка за счет сосудистого компонента. При ультразвуковом исследовании почки нормальных размеров (правая 103×46 мм, левая 95×47 мм, толщина паренхимы 19–20 мм рт. ст.), контуры ровные, эхогенность паренхимы повышена, конкрементов не выявлено. Для исключения вазоренального генеза артериальной гипертензии и почечной недостаточности была выполнена УЗДГ почечных сосудов, не выявившая признаков их окклюзии. При иммунологическом исследовании данных за системное заболевание не получено (СОЭ 13 мм/час, антинуклеарный фактор (АНФ) 0,3 ед/мл (норма до 1,0), С3 компонент комплемента 1,14 ед/мл (норма 0,9–1,8), С4–0,28 ед/мл (норма 0,1–0,4) антитела к одноцепочечной (ss) ДНК 9 ед/мл (норма до 20 ед/мл), антитела к двухцепочечной (ds) ДНК 13,7 ед/мл (норма до 20 ед/мл), антинейтрофильные цитоплазматические антитела (ANCA) — не обнаружены).
Иммунологические маркеры АФС (антитела к кардиолипину (АКЛ), волчаночный антикоагулянт (ВА)) — отрицательные. По данным коагулограммы фибриноген 4,0 г/л (норма 2–4 г/л), антитромбин III — 102% (норма 80–120%), АЧТВ — 30 сек (норма 27–39 сек), протромбиновый индекс (ПТИ) — 91% (норма 80–120%), повышение уровня растворимых комплексов фибрин-мономеров (РКФМ) до 4N (норма до 4 мг%).
Для уточнения характера поражения почек выполнена пункционная биопсия. В биоптате 22 клубочка, клубочки стянуты друг к другу, 2 из них полностью склерозированы, в 4 — ишемия капиллярных петель, еще в одном клубочке капиллярные петли коллабированы, стенка их утолщена, имеются двойные контуры. Диффузный склероз интерстиция, атрофия канальцев, занимающая более 70% площади паренхимы. Неспецифическая инфильтрация интерстиция воспалительными клетками в зоне склероза. В двух артериях малого калибра — миоинтимальная пролиферация и склероз интимы; в артериолах — мукоидное набухание интимы с сужением просвета сосудов. Заключение: тромботическая микроангиопатия.
При контрольной эхокардиографии, выполненной через месяц от дебюта заболевания, выявлено уплотнение створок митрального и трикуспидального клапанов, умеренная симметричная гипертрофия миокарда левого желудочка, незначительное снижение фракции выброса (50%). Нарушение локальной сократимости: гипоакинез задненижнего, нижнего, базального, нижнеперегородочного сегментов миокарда левого желудочка. Незначительно истончена и выбухает в полость правого предсердия межжелудочковая перегородка, без признаков патологического шунтирования. Митральная, трикуспидальная регургитация II степени. Значительная легочная гипертензия (давление в легочной артерии 53 мм рт. ст.). Консультирован кардиологом, диагностирован заднедиафрагмальный инфаркт миокарда неизвестной давности, постинфарктный кардиосклероз.
Принимая во внимание клиническую картину заболевания, данные морфологического исследования ткани почки, отрицательные серологические маркеры АФС, было выполнено генетическое исследование, выявившее мультигенную тромбофилию, представленную сочетанием гомозиготных мутаций в генах MTHFR, FGB и гетерозиготных — в генах PAI-I, тромбоцитарного рецептора фибриногена (ITGB3).
Терапия Конкором, Энапом, Тромбо-ассом позволила стабилизировать АД на уровне 150–160/90 мм рт. ст., однако отмечалось дальнейшее прогрессирование почечной недостаточности, к октябрю 2009 г. уровень креатинина крови достиг 9 мг/дл. Диагностирована терминальная ХПН, сформирована артериовенозная фистула для планового начала лечения программным гемодиализом. Однако, принимая во внимание наличие у больного мультигенной тромбофилии, в качестве «терапии отчаяния», после консультации в клинике нефрологии, внутренних и профессиональных заболеваний им. Е. М. Тареева Первого МГМУ им. И. М. Сеченова, была предпринята попытка лечения низкомолекулярными гепаринами (Клексан в максимальной дозе 80 мг/сут), на фоне которого через 2 недели было отмечено снижение уровня креатинина до 7,7 мг/дл, исчезновение одышки, нормализация уровня АД (130/90 мм рт. ст.). В связи с улучшением самочувствия больной воздержался от начала лечения гемодиализом, терапия НМГ была продолжена. Через 3 месяца от начала лечения НМГ отмечено снижение уровня креатинина до 5,1 мг/дл, еще через 2 месяца (март 2010 г.) его уровень составил 4,0 мг/дл. В результате антикоагулянтной терапии (Фраксипарин — 0,3 мл/сут), проводимой без перерыва в течение последующих 1,5 лет, удалось добиться стабилизации функции почек. При контрольном обследовании в клинике имени Е. М. Тареева в феврале 2011 г. креатинин крови сохранялся на уровне 4,0–4,5 мг/дл, D-димер — в пределах нормы, АД контролировалось на цифрах 130–140/90 мм рт. ст. приемом Лозапа 50 мг/сут и амлодипина 10 мг/сут. По данным эхокардиографии отмечена положительная динамика в виде уменьшения размеров левого и правого предсердий, нормализация фракции выброса до 56%, некоторое уменьшение выраженности легочной гипертензии (давление в легочной артерии 51 мм рт. ст.). При ультразвуковом исследовании — размеры почек несколько уменьшены, но сохраняются в пределах нормы (10×5,0 см), толщина паренхимы 16 мм, контуры четкие, неровные.
Таким образом, приведенное клиническое наблюдение иллюстрирует развитие у больного с изолированной наследственной тромбофилией мультиорганного поражения, манифестировавшего сердечной, легочной и почечной недостаточностью. Наличие полиорганной патологии в сочетании с тромбоцитопенией и микроангиопатической гемолитической анемией (МАГА), о чем свидетельствовало падение уровня гемоглобина с параллельным нарастанием уровня ЛДГ, подтвержденная морфологически ренальная тромботическая микроангиопатия позволяли обсуждать возможность развития у больного катастрофического антифосфолипидного синдрома, тромботической тромбоцитопенической пурпуры (ТТП) или атипичного гемолитико-уремического синдрома (аГУС). Однако отрицательные серологические маркеры АФС при повторных определениях позволили отвергнуть диагноз катастрофического антифосфолипидного синдрома. Умеренная тромбоцитопения, нормальный уровень ADAMTS-13 (85%), отсутствие поражения головного мозга противоречили диагнозу ТТП. Мы не имели возможности исследовать уровень факторов H и I комплемента, дефицит которых в настоящее время считается основной причиной развития аГУС, однако общая гемолитическая активность комплемента, а также уровни С3 и С4 компонентов комплемента оставались в норме. Принимая во внимание выраженную активацию внутрисосудистого свертывания крови, данные морфологического исследования ткани почки, была предпринята попытка проведения антикоагулянтной терапии, которая позволила добиться снижения креатинина до 4,5 мг/дл, АД до 130–140/90 мм рт. ст., сохранять их на достигнутом уровне в течение 2 лет и, следовательно, отсрочить начало заместительной почечной терапии.
В обоих представленных случаях не вызывает сомнение развитие ТМА: в первом случае — с изолированным поражением сосудов почек, во втором в рамках генерализованного мультиорганного поражения. По-видимому, выраженность МАГА и тромбоцитопении, являющихся диагностическими критериями классических форм ТМА (ГУС/ТТП), зависит от обширности поражения микроциркуляторного русла. Поэтому локальное поражение почечного сосудистого русла в первом случае не привело к явному формированию этих состояний. Во втором же наблюдении, напротив, отмечалось мультиорганное повреждение с вовлечением большой площади микроциркуляторного русла, что и сопровождалось развитием массивного микроангиопатического гемолиза, подтвержденного лабораторно (анемия, повышение ЛДГ), и тромбоцитопении.
Обычно органом-мишенью микроангиопатического тромбообразования при ТМА служат почки, однако в ряде случаев, как в нашем втором наблюдении, возможна генерализация процесса, приводящая к развитию полиорганной ишемии и недостаточности. В качестве причины подобных состояний в последнее время широко обсуждается в том числе и развитие аГУС взрослых. Основу этой патологии составляет дефект регуляторных механизмов системы комплемента, приводящий к неконтролируемой активации его конечных компонентов (комплекс мембранной атаки), что сопровождается повреждением эндотелия и тромбообразованием в сосудах микроциркуляторного русла жизненно важных органов.
При этом в обоих описанных случаях имеются генетические отклонения, что позволяет обсуждать имеющиеся наследственные формы тромбофилии в качестве важного этиологического фактора ТМА. В последние годы появились описания ТМА при наследственных тромбофилиях [9–14]. И если в первом наблюдении можно утверждать, что мультигенная тромбофилия явилась основной причиной почечной ТМА, то во втором — ее, скорее всего, можно рассматривать как дополнительный фактор риска развития аГУС, возможно, обеспечившей тяжесть и распространенность органного поражения. Несмотря на отсутствие четких лабораторных признаков заболевания, диагноз аГУС у второго больного может обсуждаться, поскольку патология системы комплемента, по современным представлениям, выявляется лишь у 50% больных с этой формой ТМА [15].
Представленные наблюдения демонстрируют необходимость активного диагностического поиска в отношении ТМА при сочетании признаков сосудистой нефропатии (небольшая протеинурия в сочетании со скудным мочевым осадком, тяжелая или злокачественная АГ, быстрое нарастание почечной недостаточности с признаками нарушения почечного кровотока при УЗДГ) с МАГА (шизоцитоз, повышение ЛДГ) и/или тромбоцитопенией. Снижение в этой ситуации общей гемолитической активности комплемента должно насторожить лечащего врача в отношении аГУС, диагноза, до настоящего времени крайне редко устанавливаемого взрослым пациентам. Особенности течения болезни у наших больных, в том числе и ответ на антитромботическую терапию, позволяют предполагать, что, кроме острых форм ТМА, к которым принадлежат постдиарейный и атипичный ГУС, ТТП и катастрофический АФС, возможно, по-видимому, и хроническое течение ТМА, обусловленной мультигенной тромбофилией. Это не противоречит современной классификации ТМА, где существует рубрика «обусловленная другими генетическими причинами» [2]. По-видимому, начальным признаком такой формы ТМА служит тяжелая АГ, как это имело место у обоих больных, причем у второго пациента она практически на год опередила развитие других симптомов. Можно предположить, что пребывание в жарком климате способствовало генерализации микроангиопатического тромбообразования и явилось разрешающим фактором, приведшим к развитию аГУС у больного с генетической формой тромбофилии и, возможно, патологией комплемента. Очевидна необходимость обязательного проведения в подобных случаях биопсии почки с иммуногистохимическим и электронно-микроскопическим исследованиями, позволяющими выявить как признаки острых тромбозов (около 30% случаев), представлявшихся ранее наиболее характерным морфологическим проявлением поражения почек у больных ТМА, так и, чаще, сочетания хронических изменений артерий в виде фиброзной гиперплазии интимы со сморщиванием или удвоением БМК. Подобные изменения описаны в биоптатах почек больных первичным АФС, содержавших фибриновые тромбы, и в случае такого сочетания с очевидностью указывают на существование хронической ТМА [11]. У обоих представленных пациентов имелись ее признаки — у первой больной в виде картины, напоминающей МКГН, у второго — в виде тяжелых ишемических изменений с коллапсом клубочков. Обязательным компонентом гистологической картины хронической ТМА являются изменения канальцев вплоть до тиреодизации и атрофии, фиброз интерстиция, как это отмечено у обоих пациентов. При хронической ТМА любой этиологии показано назначение противотромботической терапии. В обоих представленных нами случаях лечение низкомолекулярными гепаринами привело к улучшению функции почек: в первом — с полным восстановлением, а во втором — со значительным улучшением, позволившим отсрочить начало заместительной почечной терапии.
В последние десятилетия повсеместно в мире отмечается значительный рост числа венозных и артериальных тромбозов. Признание роли тромбофилии в развитии тромботических осложнений различных заболеваний является одним из важнейших открытий последних лет. Благодаря активным исследованиям в этой области выделены группы пациентов высокого риска в отношении развития тромбозов. К ним относятся: генетическая тромбофилия (дефицит антитромбина III, протеинов С и S, наличие лейденской мутации, мутации гена протромбина), АФС, рецидивирующие тромбозы в анамнезе, тромбоэмболические осложнения во время беременности у женщин. Однако очевидна и возможность развития тромбозов микроциркуляторного русла различных органов, в первую очередь почек, у пациентов с тромбофилиями. Следует отметить, что к тромбообразованию в сосудах малого калибра приводит, скорее, не дефицит естественных антикоагулянтов, а комбинация полиморфизмов нескольких генов гемостаза, в первую очередь PAI-I, FGB, MTHFR, как это оказалось у представленных пациентов. При сочетании тромбофилии с заболеванием почек отмечается нарастание тяжести почечного процесса с ускоренным развитием почечной недостаточности, поэтому такие состояния нуждаются в своевременной диагностике и лечении. Однако до настоящего времени в подавляющем большинстве случаев развитие острой почечной недостаточности у больных терапевтического профиля рассматривается либо как БПГН, либо как обострение ХГН, своевременно не диагностированного ранее. В случае явного цитопенического синдрома (анемия, тромбоцитопения), как правило, диагностируют системную красную волчанку с поражением почек, а при полиорганной недостаточности — системные васкулиты. По-видимому, пришло время включать в круг диагностического поиска при подобных ситуациях ТМА, в первую очередь аГУС. Это обусловлено особенностями течения ТМА, быстро приводящей к неблагоприятному исходу даже при хронической ее форме. Вот почему своевременно установленный диагноз, от которого зависит тактика лечения, может изменить прогноз.
Поводом для исключения ТМА, таким образом, у больных с поражением почек могут служить следующие признаки: остронефритический синдром в сочетании с анемией и/или тромбоцитопенией, развившаяся de novo тяжелая или злокачественная АГ или внезапное озлокачествление существовавшей ранее АГ, сочетание тяжелой АГ с нарушением функции почек независимо от наличия или отсутствия мочевого синдрома у молодого пациента, наличие указаний в анамнезе на перенесенную раннюю (до 34 нед беременности) преэклампсию, после которой в течение не менее 6 мес сохраняются АГ, нарушение функции почек, мочевой синдром — изолированные или в разных сочетаниях. Представление о тромбофилии и ТМА должно прочно войти в нефрологическую практику наряду с БПГН — идиопатическим или обусловленным системными заболеваниями.
Литература
- Levine J. S., Rauch J. Renal involvement in the antiphospholipid syndrome. In: Rhematology and the kidney. Ed. by D. Adu, P. Emery, M. Madaio. Oxford University Press, 2001: 133–166: Moake J. L. Thrombotic microangiopathies // N Engl J Med. 2002: 347: 589–600.
- Besbas N., Karpman D., Landau D. et al. A classification of hemolytic uremic syndrome and thrombotic thrombocytopenic purpura and related disorders // Kidney Intern. 2006: 70: 423–431.
- Raife T. J., Lentz S. R., Atkinson B. S. et al. Factor V Leiden: a genetic risk factor for thrombotic microangiopathy in patients with normal von Willebrand factor-cleaving protease activity // Blood. 2002. Vol. 99. P. 437–442.
- Gong R., Liu Z., Chen Z., Li L. Genetic variations in plasminogen activator inhibitor-1 gene and beta fibrinogen gene associated with glomerular microthrombosis in lupus nephritis and the gene dosage effect // Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 2002. Vol. 19. P. 1–5.
- Sucker C., Kurschat C., Farokhzad F. et al. The TT Genotype of the C677 T Polymorphism in the Methylentetrahydrofolate Reductase as a Risk Factor in Thrombotic Microangiopathies // Results From a Pilot Study Clin Appl Thromb Hemost. 2009. Vol. 15. P. 283–289.
- Боброва Л. А., Козловская Н. Л., Шкарупо В. В. и соавт. Влияние генетической формы тромбофилии на клинико-морфологические проявления и характер течения хронического гломерулонефрита // Нефрология и диализ. 2010. № 1 (12). С. 25–33.
- Боброва Л. А., Козловская Н. Л., Шкарупо В. В. и соавт. Влияние генетической формы тромбофилии на клинико-морфологические проявления и характер течения хронического гломерулонефрита // Нефрология и диализ. 2010. № 1 (12). С. 25–33.
- Боброва Л. А. Поражение почек при наследственных и приобретенных тромбофилиях. Автореф. дисс. кан. мед. наук. М., 2010.
- Raife T. J., Lentz S. R., Atkinson B. S. et al. Factor V Leiden: a genetic risk factor for thrombotic microangiopathy in patients with normal von Willebrand factor-cleaving protease activity // Blood. 2002: 99: 437–442.
- Gong R., Liu Z., Li L. Epistatic effect of plasminogen activator inhibitorI and β-fibrinogen genes on risk of glomerular microthrombosis in lupus nephritis // Arthritis Rheum 2007: 56: 1608–1617, доказана принадлежность к группе ТМА АФС-нефропатии.
- Nochy D. Daugas E., Droz D. et al. The Intrarenal Vascular Lesions Associated with Primary Antiphospholipid Syndrome // J Am Soc Nephrol. 1999: 10: 507–518.
- Шилов Е. М., Козловская Н. Л., Метелева Н. А. и соавт. Клинические проявления нефропатии, связанной с антифосфолипидным синдромом, при первичном антифосфолипидном синдроме // Терапевт. архив. 2003: 75 (6): 22–27.
- Козловская Н. Л., Шилов Е. М. Нефропатия при антифосфолипидном синдроме как вариант тромботического микроангиопатического поражения почек // Российский медицинский журнал. 2002, № 4, с. 3–5.
- Piette J. C., Cacoub P., Wechsler B. Renal manifestation of the antiphospholipid syndrome. Semin Arthritis Rheum 1994: 23: 357–366.
- Hirt-Minkowski P., Dickenmann M., Schifferli J. A. Atypical hemolytic uremic syndrome: update on the complement system and what is new // Nephron Clin Pract. 2010: 114 (4): c219–35.
Н. Л. Козловская*, доктор медицинских наук, профессор
И. Б. Колина*, **, кандидат медицинских наук
Л. А. Боброва*, **, кандидат медицинских наук
А. М. Кучиева*
Е. Ю. Хафизова*
*ГБОУ ВПО Первый МГМУ им. И. М. Сеченова Минздравсоцразвития России,
**НИИ уронефрологии и репродуктивного здоровья человека, Москва
Контактная информация об авторах для переписки: ikolina@yandex.ru
Купить номер с этой статьей в pdf